Generalidade
A esclerose tuberosa é uma doença genética que afeta vários órgãos e tecidos do corpo humano. Por esse motivo, apresenta um amplo espectro de sintomas, alguns típicos da primeira infância, outros da idade adulta.A esclerose tuberosa pode ser transmitida dos pais para os filhos, mas também pode surgir devido a uma mutação espontânea do DNA.

O que é esclerose tuberosa
A esclerose tuberosa é uma doença genética caracterizada pela formação de hamartomi em diferentes órgãos ou tecidos.
Hamartoma identifica uma área de tecido onde as células se multiplicaram intensamente, formando uma massa perceptível, semelhante a um caroço ou tubérculo. Os hamartomas são uma reminiscência de tumores, mas não devem ser confundidos com eles: na verdade, as células do hamartoma são idênticas às do tecido em que proliferam; as de um tumor, por outro lado, têm características diferentes. E dão aumento de neoplasias benignas, miomas e angiofibromas.
O cérebro, a pele, os rins, os olhos, o coração e os pulmões são as áreas mais afetadas, mas não são os únicos locais. Devido à multiplicidade de órgãos e tecidos envolvidos, a esclerose tuberosa também é definida como uma doença genética multissistêmica.
Mais tarde, será compreendido por que os hamartomas aparecem apenas em certas áreas.
Epidemiologia
Os dados sobre a incidência e o número de casos em todo o mundo são incertos, mas a incerteza se deve ao fato de que muitos pacientes não apresentam sintomas e levam uma vida normal.
No entanto, estima-se que a incidência de esclerose tuberosa seja de um caso para cada 5.000-10.000 novos nascimentos, ocorrendo cerca de dois milhões de casos em todo o mundo.
Causa
A esclerose tuberosa é uma doença genética; isso significa que um gene, presente no DNA da pessoa afetada, sofre mutação.
Os genes que, quando afetados pelas mutações relativas, causam esclerose tuberosa são dois:
- TSC1.
- TSC2.
Os casos de esclerose tuberosa observados até agora apresentam apenas um desses genes mutado. Portanto, a única mutação de TSC1, ou de TSC2, é suficiente para causar esclerose tuberosa.
Estudos realizados na Europa e nos Estados Unidos relatam que a mutação em TSC2 (80% dos casos) é muito mais frequente do que em TSC1 (os 20% restantes).
TSC1 E TSC2
O gene TSC1 reside no cromossomo 9 e produz uma proteína chamada hamartina.
O gene TSC2 reside no cromossomo 19 e produz uma proteína chamada tuberina.
As proteínas produzidas, hamartina e tuberina, unem-se e funcionam juntas. Isso explica por que a mutação de um ou de outro causa a mesma patologia.
FUNÇÃO DE TSC1 E TSC2
Eles são considerados genes supressores de tumor e desempenham um papel fundamental nos processos de:
- Crescimento e diferenciação celular durante a embriogênese.
- Síntese proteíca.
- Autofagia.
Quando TSC1 e TSC2 são mutados, as proteínas produzidas são defeituosas e esses processos fisiológicos não ocorrem mais regularmente.
Crescimento e diferenciação celular durante a embriogênese
Síntese proteíca
Autofagia
Crescimento e diferenciação celular durante a embriogênese
Síntese proteíca
Autofagia
INÍCIO DOS AMARTOMAS
Os hamartomas podem surgir quando ocorre uma mutação em um gene que controla o crescimento e a diferenciação celular, como TSC1 ou TSC2. As células, conseqüentemente, crescem em número, gerando massas evidentes; deste modo, formam-se placas de formato semelhante a um nódulo ou tubérculo. Na histologia, esse processo é definido pelo termo hiperplasia.
GENÉTICA
Duas premissas:
- Cada gene de DNA humano está presente em duas cópias. Essas cópias são chamadas de alelos.
- O ser humano possui 23 pares de cromossomos, destes, apenas um par determina o sexo (cromossomos sexuais), todos os demais são chamados de cromossomos autossômicos.
A esclerose tuberosa é uma doença genética autossômica dominante. Para isso, basta que um alelo seja mutado para que todo o gene não funcione adequadamente. O alelo mutado, de fato, tem mais poder do que o saudável (domínio).
Na verdade, os distúrbios da esclerose tuberosa são agravados quando os alelos TSC1 ou TSC2 sofrem mutação. Ou seja, apenas um alelo, mesmo que dominante sobre o outro, não causa sintomas evidentes, nestes casos falamos de alelos com dominância incompleta.
HERANÇA € OU MUTAÇÃO ESPONTÂNEA?
A mutação TSC1 ou TSC2 pode surgir de:
- Transmissão hereditária (ou seja, de um dos pais) de um alelo mutado.
- Mutação espontânea de um alelo no estágio embrionário (ou embriogênese).
Um terço dos casos de esclerose tuberosa é devido à transmissão hereditária. Nestes casos, basta que um dos pais tenha uma mutação nos genes TSC1 ou TSC2 para que a prole seja afetada pela doença (vimos de fato que a esclerose tuberosa é uma doença hereditária autossômica dominante).
Os restantes 2/3 dos casos são devidos a uma mutação espontânea durante a fase embrionária.
TSC1 em 50%
TSC2 nos restantes 50%
TSC2 em 70%
TSC1 em 30%
POR QUE SÓ CERTOS ÓRGÃOS SÃO AFETADOS?
Premissa: o embrião, durante os primeiros estágios de seu desenvolvimento, possui três camadas de células:
- Ectoderma, o mais externo.
- Mesoderma, a central.
- Endoderma, o mais íntimo.
Órgãos e tecidos específicos derivam de cada camada.
Sistema nervoso
Epiderme
Epitélio da boca
Epitélio do cólon
Tesão e cristalino
Esmalte dentário
Ossos dérmicos
Coração
Rim
Forro da parede intestinal
Musculatura dos membros
Membranas serosas dos pulmões (pleura) e do coração (pericárdio).
Fígado
Pâncreas
Sistema digestivo
Agora temos todos os elementos para entender por que os hamartomas surgem apenas em certas partes do corpo.
Mutações de TSC1 ou TSC2 ocorrem no estágio embrionário nas células do ectoderma e mesoderma. Portanto, os tecidos que irão surgir dessas camadas celulares apresentarão hamartomas.
Sintomas
Para mais informações: Esclerose Tuberosa - Causas e Sintomas
Os órgãos e tecidos afetados pela esclerose tuberosa são numerosos. Os distritos mais afetados são:
- Cérebro, pele, rins, coração, olhos
Mas outras doenças mais raras não devem ser esquecidas, em detrimento de:
- Pulmões, intestinos, fígado, dentes, sistema endócrino, ossos
Alguns sintomas aparecem em uma idade jovem, outros na idade adulta.
DOMINÂNCIA INCOMPLETA
Já foi mencionado acima que a dominância do alelo mutado dos genes TSC1 ou TSC2 é incompleta, o que significa que o alelo saudável ainda é capaz de produzir uma proteína "saudável" (hamartina ou tuberina), embora em quantidade inferior. A presença da proteína "saudável" compensa os danos causados pela proteína mutada. Nessas condições, os hamartomas ainda não causam manifestações dramáticas.
Quando o outro alelo também muda (este é um evento raro, mas possível), os hamartomas crescem de forma descontrolada.
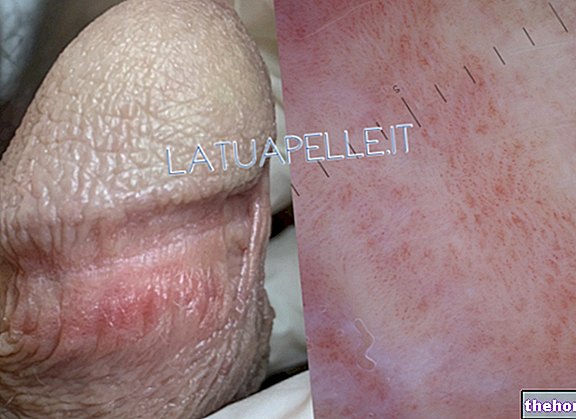
MANIFESTAÇÕES DE PELE
Cerca de 90% dos pacientes apresentam alterações cutâneas. Os eventos são numerosos e variados. Os típicos são manchas despigmentadas, adenomas sebáceos de Pringle e tumores de unha de Koenen.
Manchas despigmentadas são manchas hipomelanóticas, ou seja, com menor teor de melanina
Os adenomas sebáceos Pringle são tumores benignos também chamados de angiofibromas faciais. Os hamartomas aparecem como pequenas massas globulares e vermelhas. Os tumores das unhas de Koenen são miomas e surgem de hamartomas de alguns milímetros.
Fotos na pele com manifestações de esclerose tuberosa
A tabela mostra as inúmeras manifestações cutâneas devido à esclerose tuberosa:
Tronco
Artes
Bochechas
Nariz
Queixo
Unhas e mãos
Frente
Seu couro cabeludo
Tronco
Região dorso-lombar
Pescoço
Ombros
Dentes
Boca
Goma anterior
Lábio
Palato
SINTOMAS NEUROLÓGICOS
Os locais do cérebro afetados pela esclerose tuberosa são:
- O córtex cerebral
- A matéria branca
- Os ventrículos
- Os gânglios da base
As duas figuras ajudam o leitor a entender as áreas afetadas.

Dependendo da localização e da forma dos hamartomas, diferentes distúrbios podem ocorrer, tais como:
- Epilepsia
- Nódulos subependimários
- Tumores cerebrais do tipo astrocitoma
- Déficits mentais, comportamentais e de aprendizagem.
Tubérculo
Latido
80-90%
- Espasmos
- Parcial
- Febril
Primeira infância (espasmos), 75%
Idade adulta (parcial), 25%
Nódulo
Ventrículos
80-90%
Infância
Hidrocefalia obstrutiva
Evolução para astrocitoma subpendimal
Cistos cerebrais
Nódulo
> 1 cm
Ventrículos (Foramina di Monro)
6%
Entre 4 e 10 anos
Dor de cabeça
Ele vomitou
Convulsões
Alterações do campo visual
Mudanças repentinas de humor
Hidrocefalia
Cistos cerebrais
Deficiência mental
Primeira infancia
(0-5 anos)
Requer supervisão (85%)
Ausência de linguagem (65%)
Não autossuficiente (60%)
Autismo
Déficit de atenção
Hiperatividade
Agressão
Auto-mutilação
Distúrbios do sono
Infância
Associação com epilepsia
Família difícil e gestão escolar
LESÕES RENAS
Eles são muito frequentes. Na verdade, eles aparecem em 60-80% dos casos. Eles consistem em:
- Hamartomas semelhantes a tumores benignos.
- Malformações da estrutura renal.
Angiomiolipoma (60-70%)
Angiolipoma
Miolipomas
Eles são tumores benignos, que aparecem em várias formas
Durante a infância: assintomático
Na idade adulta: possível ruptura do hamartoma, seguida de hemorragia, hematúria e dor abdominal.
Falência renal
Rim em ferradura
Rim policístico
Ausência de um rim (agenesia renal)
Ureter duplo
LESÕES CARDIOVASCULARES
Novamente, eles são devidos a hamartomas semelhantes a tumores benignos, chamados de rabdomiomas.
Assintomático.
Se as dimensões forem grandes:Arritmias
Mudanças no fluxo cardíaco
LUNGES PULMONARES
Devem-se principalmente à linfangioleiomiomatose pulmonar (LMA) e, em menor grau, à hiperplasia multifocal micronodular. Eles são manifestações típicas da idade adulta.
Doença rara
Afeta principalmente mulheres adultas
Cistos pulmonares aparecem
A maioria dos casos são assintomáticos
Os sintomas são: dispneia semelhante à asma, tosse, pneumotórax espontâneo, insuficiência respiratória
Doença rara
Afeta principalmente adultos, homens e mulheres
Nódulos aparecem, visíveis em uma radiografia de tórax
Quase sempre assintomático
OUTRAS LESÕES
Hamartoma retinal
Astrocitoma retinal
Pólipos intestinais
Cistos intestinais
Angiomiolipoma
Angiomas
Pseudo-cisto nas mãos e pés
Adenomas
Angiomiolipomas
Diagnóstico
O diagnóstico consiste em:
- Anamnese
- Análise clínica dos sinais acima mencionados
- Exames instrumentais
ANAMNESE
O médico faz um “levantamento da história familiar do paciente, para saber se a esclerose tuberosa é hereditária ou por mutação espontânea.
ANÁLISE CLÍNICA DOS SINAIS
Em 1998, um grupo de médicos internacionais estabeleceu um critério diagnóstico com base nas manifestações clínicas mencionadas. Eles foram divididos em:
- Principais sinais (ou critérios)
- Sinais menores (ou critérios)
Se o paciente mostra
- 2 sinais principais,
- 1 sinalizador principal e 2 sinais secundários
Se o paciente mostra
- 1 sinal principal
- 2 ou mais sinais menores
A classificação dos sinais é a seguinte:
EXAMES INSTRUMENTAIS
Tomografia computadorizada do cérebro
Ressonância magnética nuclear
- Tubérculos do córtex cerebral
- Nódulos subependimários
- Astrocitomas de células gigantes subependimárias (SEGA)
Sim (radiação ionizante)
Não
Espirometria
Raio-x do tórax
- Linfangioleiomiomatose pulmonar
- Parada respiratória
Não
Sim (radiação ionizante)
TESTE GENÉTICO
É uma investigação longa, que demora alguns meses, pelo que não é útil para o diagnóstico precoce, mas sim para confirmar o diagnóstico com base nos sinais clínicos.
Terapia
Não há cura específica e eficaz, já que a esclerose tuberosa é uma:
- Doença genética.
- Doença multissistêmica.
No entanto, alguns sintomas podem ser controlados para evitar complicações e melhorar a qualidade de vida dos pacientes.
TRATAMENTO FARMACOLÓGICO
As manifestações clínicas que podem ser tratadas com a administração de medicamentos são:
- Epilepsia infantil
- Linfangioleiomiomatose pulmonar (LAM)
- Doenças renais
Epilepsia infantil. O pequeno paciente recebe medicamentos anticonvulsivos:
- ACTH (hormônio adrenocorticotrópico)
- Vigabatrina
Linfangioleiomiomatose pulmonar. Os broncodilatadores do tipo beta-2 agonista, como o salbutamol, são úteis. No entanto, a eficácia da terapia hormonal baseada em progesterona ou buserelina é incerta
Doenças renais. Anti-hipertensivos, como inibidores da ECA e diuréticos, são usados.
TRATAMENTOS FÍSICO-CIRÚRGICOS
Eles consistem em intervenções destinadas a remover:
- Angiofibromas faciais
- Miomas de unha
- As placas de pele
- Os pontos serrilhados
- Astrocitomas de células gigantes subependimárias (SEGA)
- Angiomiolipomas renais
- Lesões pulmonares
- Os tubérculos do córtex cerebral, que causam epilepsia
A tabela a seguir resume os principais tratamentos terapêuticos e suas características.
Diatermia
Crioterapia
Remoção cirúrgica
Minimamente invasivo
sim
Terapia a laser
Remoção cirúrgica
sim
Acompanhamento e prognóstico
Introdução: o termo médico acompanhamento refere-se ao paciente que, portador de câncer, foi submetido positivamente à cirurgia.
Verificações periódicas são recomendadas para acompanhamentos. A oftalmoscopia, ou seja, o exame de fundo de olho, também pode ser realizada uma vez por ano. Por outro lado, as doenças neurológicas, cardíacas e renais requerem monitoramento mais frequente.
PROGNÓSTICO
A evolução da esclerose tuberosa é variável e depende de caso para caso.
Alguns pacientes apresentam sintomas leves, quase imperceptíveis. Para estes, a qualidade de vida não é afetada pela doença e o prognóstico é excelente.
Por outro lado, outros pacientes apresentam sintomas muito mais dramáticos e evidentes. A morte ocorre principalmente por lesões neurológicas, portanto, o prognóstico torna-se muito desfavorável.
CONSULTORIA GENÉTICA
Se um dos pais tiver esclerose tuberosa, a probabilidade de um filho herdar a mesma condição é de 50%.
Se, por outro lado, um filho de pais saudáveis for afetado, a probabilidade de um segundo filho ficar doente é muito baixa. Nestes casos, um teste genético esclarece se os pais são portadores de esclerose tuberosa ou se, em vez disso, ocorreu uma mutação espontânea.



-cos-cause-e-terapia.jpg)