Esta doença deve o seu nome ao endocrinologista americano que a descobriu: Frederic Crosby Bartter. A incidência anual foi estimada em 1 / 830.000.
Existem várias variantes da síndrome de Bartter cuja transmissão, embora ainda autossômica, pode variar de recessiva a dominante, dependendo do caso.
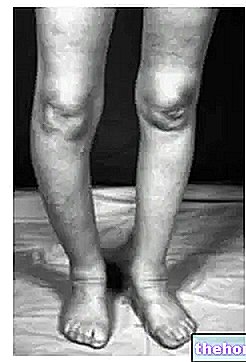
Se não for diagnosticada e tratada prontamente, a síndrome de Bartter pode comprometer gravemente o desenvolvimento, o crescimento e a qualidade de vida do paciente. Além disso, em casos particularmente graves, a expectativa de vida é consideravelmente reduzida.
Observe
A síndrome de Bartter NÃO deve ser confundida com a síndrome de Schwartz-Bartter, uma doença caracterizada por "secreção prejudicada do hormônio antiduirético (ADH), também conhecida como síndrome da secreção inadequada de ADH (SIADH).
.que ocorre ao nível da alça de Henle é atribuível a uma "alteração da síntese de alguns receptores de canal / transportador (proteínas particulares que transportam íons de natureza diferente) localizados nesta" área do rim. Esse fenômeno é causado por uma série de mutações genéticas que afetam os genes que codificam as proteínas específicas acima mencionadas.
As diferentes variantes da síndrome de Bartter são diferenciadas de acordo com o gene afetado. Informações mais detalhadas sobre isso podem ser encontradas no capítulo a seguir.
.
A tabela a seguir mostrará, portanto, as diferentes variantes da síndrome, os genes mutantes envolvidos, as proteínas (receptores / transportadores de canal) para as quais codificam e a apresentação clínica da variante em questão.
Variante
Gene mutado
Canal / transportador envolvido
Apresentação clínica
Síndrome de Bartter tipo I
Gene SLC12A1
NKCC2 (cotransportador sódio-potássio-cloro ou Na + / K + / 2Cl-)
Síndrome de Bartter pré-natal (ou infantil)
Síndrome de Bartter tipo II
Gene KCNJ1
ROMK (canal de potássio da medula externa do rim)
Síndrome de Bartter pré-natal (ou infantil)
Síndrome de Bartter tipo III
Gene CLNKb
CLCNKb (canal de cloro tipo Kb)
Síndrome de Bartter Clássica
Síndrome de Bartter tipo IV ou IV A
Gene BSND
Barttina (subunidade beta dos canais de cloro do tipo Ka e Kb)
Síndrome de Bartter pré-natal (ou infantil) e perda auditiva neurossensorial
Síndrome de Bartter tipo IV B
Genes CLCNKa e CLCNKb
CLCNKa (canal de cloro tipo Ka) e CLCNKb
Síndrome de Bartter pré-natal (ou infantil) e surdez neurossensorial
Síndrome de Bartter tipo V
Gene CASR
CaSR (receptor sensível ao cálcio)
Síndrome de Bartter com hipocalcemia
Como pode ser visto na tabela, apesar da presença de cinco variantes genéticas, não é possível distinguir tantas formas clínicas; na verdade, apenas quatro são distintos: síndrome de Bartter pré-natal ou infantil (tipo I e II), síndrome de Bartter clássica (tipo III), síndrome de Bartter pré-natal ou infantil associada à surdez neurossensorial (tipo IV A e IV B; algumas fontes, no entanto, eles agrupam essas variantes com os tipos I e II) e, finalmente, a síndrome de Bartter com hipocalcemia (tipo V).
Você sabia disso ...
Dada a existência de uma variante IV (ou IV A) e uma variante IV B da síndrome de Bartter, algumas fontes consideram, em geral, seis variantes da síndrome de Bartter.Outras fontes, no entanto, consideram a variante IV B como um subtipo da variante IV e, por esse motivo, contemplam a existência de apenas cinco variantes genéticas da síndrome de Bartter.
As variantes do tipo I, II, III, IV e IV B são doenças autossômicas de transmissão recessiva, o que significa que para manifestar a síndrome, o indivíduo deve possuir ambos os alelos mutados herdados dos pais que, portanto, serão portadores saudáveis. da síndrome, por outro lado, é uma doença de transmissão autossômica dominante, ou seja, para manifestar os sintomas, basta que o paciente possua um único alelo mutado que, portanto, pode ser herdado até mesmo por um (também doente ) dos dois pais.
Pseudo-síndrome de Bartter
A pseudo-síndrome de Bartter é uma condição caracterizada por sintomas semelhantes aos induzidos pela síndrome de Bartter, mas cuja causa pode ser encontrada no abuso de drogas diuréticas como a furosemida.
Síndrome de Gitelman
Esta síndrome é causada por uma mutação localizada no gene SLC12A3 que codifica o co-transportador sódio-cloro (NCC). Devido a esta mutação - transmitida de forma autossômica recessiva - o paciente sofre um comprometimento da reabsorção de sódio, cloro e potássio no nível do túbulo contorcido distal, ao contrário da síndrome de Bartter em que o comprometimento da reabsorção está localizado no "No entanto , A síndrome de Gitelman pode originar sintomas semelhantes aos da síndrome de Bartter, razão pela qual, na prática clínica, às vezes pode ser difícil distinguir as duas doenças.
, hipocloremia e alcalose metabólica que podem estar associadas a hiperreninemia (renina alta no sangue) e hiperaldosteronismo. É claro que todas essas condições podem, por sua vez, dar origem a uma série de sintomas capazes de comprometer a qualidade de vida do paciente (por exemplo, náusea, vômito, tontura, fraqueza, dor de cabeça, hipotensão, etc.).
Além do que foi dito até agora, cada variante pode dar origem a manifestações e sintomas específicos intimamente relacionados ao gene mutado e ao consequente envolvimento do canal ou cotransportador para o qual esse gene codifica. Portanto, os sintomas e manifestações típicos associados a cada uma das cinco formas diferentes da síndrome de Bartter serão brevemente descritos a seguir.
Síndrome de Bartter tipo I
Na síndrome de Bartter tipo I, as mutações afetam o gene que codifica o cotransportador sódio-potássio-cloro presente na alça de Henle, pois devido ao comprometimento da reabsorção ocorre hipovolemia por perda de sais. Ao mesmo tempo, como a reabsorção de cálcio também está ligada à atividade do referido cotransportador, assistimos ao aparecimento de hipercalciúria, o que pode levar ao aparecimento de nefrocalcinose. Também é possível ter hipermagnesúria. A polidrâmnio secundária à poliúria fetal pode se desenvolver no período pré-natal.
Síndrome de Bartter tipo II
A síndrome de Bartter tipo II é causada por uma mutação no gene que codifica o canal de potássio da medula adrenal. As manifestações e sintomas são semelhantes aos da variante I e também neste caso pode-se encontrar polidrâmnio secundário à poliúria fetal. No entanto, em um estágio inicial, o recém-nascido pode apresentar acidose metabólica hipercalêmica transitória. Essa condição evolui então para o quadro clínico característico da síndrome de Bartter.
Síndrome de Bartter tipo III
Também conhecida como síndrome de Bartter clássica, a variante III da doença é causada por mutações no gene que codifica o canal de cloro do tipo Kb. Como os canais de cloro do tipo Ka são preservados nessa forma, os sintomas tendem a ser mais leves, embora ainda presentes. Geralmente não há nefrocalcinose.
Síndrome de Bartter tipo IV e IV B
Em ambos os tipos de variante IV há envolvimento de genes envolvidos na síntese correta dos canais de cloro Ka e Kb. Como ambos os canais estão comprometidos, os sintomas tendem a ser mais graves do que na variante III da síndrome. Os bebês podem apresentar inicialmente um quadro clínico que imita o hipoaldosteronismo, mas que então evolui para alcalose metabólica hipocalêmica quando o corpo tenta compensar a falta de atividade dos canais de cálcio mencionados. A característica das variantes IV e IV B da síndrome de Bartter é o aparecimento de neurossensorial surdez.
Síndrome de Bartter tipo V
A variante V da síndrome de Bartter é causada por uma mutação que afeta o gene que codifica o receptor sensível ao cálcio, envolvido na inibição da reabsorção de água e vários íons, como cálcio, potássio e sódio. O receptor leva ao aparecimento de hipocalcemia e hipercalciúria conseqüente associada aos sintomas característicos da síndrome de Bartter.
Você sabia disso ...
As variantes I, II, IV e IV B da síndrome de Bartter - bem como com o nome de síndrome de Bartter pré-natal - às vezes também são chamadas de síndrome da hiperprostaglandina E2, pois se caracterizam por um aumento nos níveis plasmáticos dessa prostaglandina.
- visa identificar a presença e concentração de eletrólitos (sódio, potássio, cloreto, magnésio, bicarbonato, cálcio) e substâncias específicas (renina e aldosterona) no plasma e / ou urina.O diagnóstico definitivo, entretanto, só é possível com a realização de testes genéticos específicos.
O diagnóstico diferencial, por outro lado, deve ser colocado contra a pseudo-síndrome de Bartter, a síndrome de Gitelman, a fibrose cística e a doença celíaca.
Nos casos em que existe um certo risco (por exemplo, pais com portadores saudáveis e / ou doentes) de o recém-nascido manifestar a doença, o diagnóstico pré-natal também é possível.
a partir de:
- Suplementos de sais minerais (em particular, mas não exclusivamente, potássio) para compensar a falta de reabsorção;
- Antiinflamatórios não esteroidais (AINEs) como, por exemplo, indometacina Esses medicamentos são administrados com o objetivo de diminuir níveis excessivamente elevados de prostaglandina E2;
- Diuréticos poupadores de potássio (administrados para reduzir a excreção de potássio na urina).
Nos casos mais graves e / ou em condições estressantes (aparecimento de outras doenças, intervenções cirúrgicas, etc.), a reposição de potássio e outros sais minerais pode ser realizada por via intravenosa, é claro, uma operação semelhante deve ser realizada pela saúde pessoal especializado.






















.jpg)