Generalidade
A anemia mediterrânea (ou talassemia beta) é uma doença hereditária do sangue.
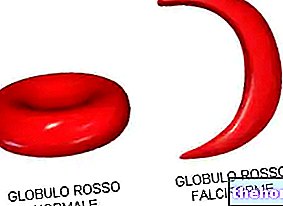
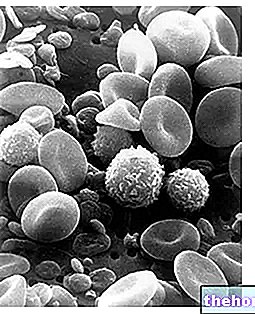
Os pacientes afetados apresentam menos glóbulos vermelhos do que o normal, com defeitos na síntese de hemoglobina (Hb, proteína responsável pelo transporte de oxigênio).

A extensão da doença, os sintomas e as consequências são muito variáveis e dependem fundamentalmente do tipo de defeito genético. Existem, de facto, 3 formas diferentes de anemia mediterrânica:
- Talassemia major (ou doença de Cooley);
- Talassemia intermediária;
- Talassemia menor.
Nos casos mais graves, a anemia mediterrânea é incapacitante e fatal, nas outras formas é quase assintomática.Há também a possibilidade de ser um portador são, com risco de ter filhos que irão desenvolver a doença.
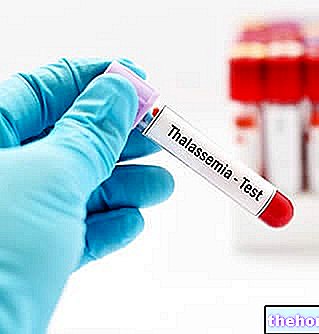
A anemia mediterrânea é detectável através de testes genéticos e de sangue, este último irá evidenciar a presença de glóbulos vermelhos de tamanho irregular, frágeis, escassos e menores que o normal.
O tratamento envolve várias abordagens, incluindo transfusões de sangue mais ou menos recorrentes associadas à terapia de quelação (para evitar o acúmulo de ferro) e transplante de medula óssea de doadores compatíveis. Às vezes, nenhuma intervenção terapêutica é necessária.
-cosa-significa-quando-preoccuparsi.jpg)