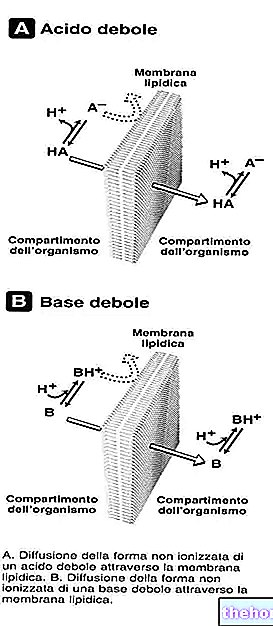
Ingredientes ativos: Levetiracetam
Comprimidos revestidos por película de Keppra 250 mg
Comprimidos revestidos por película de Keppra 500 mg
Comprimidos revestidos por película de Keppra 750 mg
Comprimidos revestidos por película de Keppra 1000 mg
As bulas de Keppra estão disponíveis para os tamanhos de embalagem: - Keppra 250 mg comprimidos revestidos por película, Keppra 500 mg comprimidos revestidos por película, Keppra 750 mg comprimidos revestidos por película, Keppra 1000 mg comprimidos revestidos por película
- Keppra 100 mg / ml solução oral
- Keppra 100 mg / ml concentrado para solução para perfusão
Por que o Keppra é usado? Para que serve?
O Keppra é um medicamento antiepiléptico (um medicamento utilizado para tratar convulsões).
Keppra é usado:
- por conta própria em adultos e adolescentes a partir de 16 anos de idade com epilepsia recém-diagnosticada, para tratar convulsões parciais com ou sem generalização secundária.
- como um complemento a outros medicamentos antiepilépticos para tratar:
- crises de início parcial, com ou sem generalização, em adultos, adolescentes, crianças e bebês a partir de 1 mês de idade
- convulsões mioclônicas em adultos e adolescentes a partir dos 12 anos de idade com epilepsia mioclônica juvenil
- convulsões tônico-clônicas generalizadas primárias em adultos e adolescentes a partir dos 12 anos de idade com epilepsia generalizada idiopática.
Contra-indicações Quando Keppra não deve ser usado
Não tome Keppra
- Se tem alergia (hipersensibilidade) ao levetiracetam ou a qualquer outro componente deste medicamento
Precauções de uso O que você precisa saber antes de tomar Keppra
Fale com o seu médico antes de tomar Keppra
- Se você tiver problemas renais, siga as instruções do seu médico. Este último pode decidir se a dose precisa ser corrigida.
- Se notar um abrandamento do crescimento ou um desenvolvimento inesperado da puberdade no seu filho, contacte o seu médico.
- Se notar um aumento na gravidade das crises (por exemplo, aumento do número), entre em contato com o seu médico.
- Um número limitado de pessoas que iniciaram o tratamento com antiepilépticos como o Keppra teve pensamentos de agressão ou suicídio. Se tiver quaisquer sintomas de depressão e / ou pensamentos suicidas, contacte o seu médico.
Interações Quais medicamentos ou alimentos podem alterar o efeito de Keppra
Outros medicamentos e Keppra
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica.
Keppra com comida, bebida e álcool
Você pode tomar Keppra com ou sem alimentos. Como medida de segurança, não tome Keppra com álcool.
Avisos É importante saber que:
Gravidez e amamentação
Consulte o seu médico ou farmacêutico antes de tomar qualquer medicamento.
Se estiver grávida ou pensar que pode estar grávida, informe o seu médico. Keppra não deve ser usado durante a gravidez, a menos que seja claramente necessário. O risco de defeitos congênitos para o feto não pode ser completamente descartado. Keppra demonstrou efeitos reprodutivos indesejáveis em estudos em animais com níveis de dose superiores aos necessários para controlar as convulsões.
A amamentação não é recomendada durante o tratamento.
Condução e utilização de máquinas
Keppra pode reduzir a capacidade de conduzir ou utilizar ferramentas ou máquinas, uma vez que Keppra pode causar sonolência. Isto é mais provável no início do tratamento ou após um aumento da dose.Não deve conduzir ou utilizar máquinas até ter verificado que a sua capacidade para realizar estas atividades não é afetada.
Os comprimidos revestidos por película de Keppra 750 mg contêm Sunset Yellow FCF (E110)
O corante Sunset Yellow FCF (E110) pode causar reações alérgicas. Outras dosagens de comprimidos de Keppra não contêm este componente.
Dose, método e tempo de administração Como usar Keppra: Posologia
Tome este medicamento sempre de acordo com as indicações do seu médico ou farmacêutico.
Em caso de dúvida, consulte o seu médico ou farmacêutico. O Keppra deve ser tomado duas vezes ao dia, uma de manhã e outra à noite, aproximadamente à mesma hora todos os dias.
Tome o número de comprimidos de acordo com as instruções do seu médico.
Monoterapia
- Dose para adultos e adolescentes (a partir dos 16 anos):
Dose típica: entre 1000 mg e 3000 mg por dia.
Quando você começar a tomar Keppra pela primeira vez, o seu médico irá prescrever uma dose mais baixa por 2 semanas antes de lhe dar a dose mais baixa típica.
Exemplo: se sua dose diária é de 1000 mg, você pode tomar 2 comprimidos de 250 mg de manhã e 2 comprimidos de 250 mg à noite.
Terapia adjuvante
- Dose para adultos e adolescentes (12 a 17 anos) com peso igual ou superior a 50 kg:
Dose típica: entre 1000 mg e 3000 mg por dia.
Exemplo: se a sua dose diária é de 1000 mg, você pode tomar 2 comprimidos de 250 mg de manhã e 2 comprimidos de 250 mg à noite.
- Dose para bebês (6 a 23 meses), crianças (2 a 11 anos) e adolescentes (12 a 17 anos) com peso inferior a 50 kg:
O seu médico irá prescrever a forma farmacêutica mais adequada de Keppra dependendo da sua idade, peso e dose.
Keppra 100 mg / ml solução oral é a apresentação mais adequada para lactentes e crianças com idade inferior a 6 anos.
Dose típica: entre 20 mg por kg de peso corporal e 60 mg por kg de peso corporal por dia.
Exemplo: Para uma dose diária típica de 20 mg por kg de peso corporal, se o seu filho pesar 25 kg, você pode dar a ele 1 comprimido de 250 mg de manhã e 1 comprimido de 250 mg à noite
- Dose para bebês (1 mês a menos de 6 meses):
Keppra 100 mg / ml solução oral é a apresentação mais adequada para crianças.
Método de administração:
Engula os comprimidos de Keppra com uma quantidade suficiente de líquido (por exemplo, um copo de água).
Duração do tratamento:
- Keppra é usado como tratamento crônico. O tratamento com Keppra deve durar o tempo que o seu médico lhe indicar.
- Não interrompa o tratamento sem o conselho do seu médico, pois isso pode aumentar o número de convulsões. Se o seu médico decidir interromper o tratamento com Keppra, ele irá instruí-lo sobre como interromper gradualmente o tratamento com Keppra.
Sobredosagem O que fazer se você tiver tomado Keppra em excesso
Se você tomar mais Keppra do que deveria:
Os possíveis efeitos colaterais de uma sobredosagem com Keppra são sonolência, agitação, agressão, diminuição do estado de alerta, inibição da respiração e coma. Contacte o seu médico se tiver tomado mais comprimidos do que deveria. O seu médico irá determinar o melhor tratamento possível para a sobredosagem.
Se você se esquecer de tomar Keppra:
Contacte o seu médico se se esqueceu de tomar uma ou mais doses. Não tome uma dose a dobrar para compensar um comprimido esquecido
Se você parar de tomar Keppra:
Em caso de descontinuação do tratamento, como com qualquer outro medicamento antiepiléptico, Keppra deve ser descontinuado gradualmente para evitar o aumento das convulsões.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
Efeitos colaterais Quais são os efeitos colaterais do Keppra
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham. Alguns dos efeitos secundários, como sonolência, cansaço e tonturas, podem ser mais frequentes no início do tratamento ou quando a dose é aumentada, mas estes efeitos devem diminuir com o tempo.
Muito comuns: podem afetar mais de 1 em 10 pacientes
- nasofaringite;
- sonolência, dor de cabeça.
Frequentes: podem afetar 1 a 10 em 100 pacientes
- anorexia (perda de apetite);
- depressão, hostilidade ou agressão, ansiedade, insônia, nervosismo ou irritabilidade;
- convulsão, distúrbio do equilíbrio, tontura (sensação de instabilidade), letargia, tremor (tremores involuntários);
- vertigem (sensação de rotação);
- tosse;
- dor abdominal, diarreia, dispepsia (indigestão), vômito, náusea;
- irritação na pele;
- astenia / fadiga (sensação de fraqueza).
Pouco frequentes: podem afetar 1 a 10 em 1000 pacientes
- diminuição do número de plaquetas no sangue, diminuição do número de glóbulos brancos;
- perda de peso, ganho de peso;
- tentativa de suicídio e ideação suicida, transtorno mental, comportamento anormal, alucinações, raiva, confusão, ataque de pânico, instabilidade emocional / alterações de humor, agitação;
- amnésia (perda de memória), comprometimento da memória (esquecimento), coordenação / ataxia anormal (comprometimento da coordenação motora), parestesia (formigamento), comprometimento da atenção (perda de concentração);
- diplopia (visão dupla), visão turva;
- teste de função hepática anormal;
- queda de cabelo, eczema, coceira;
- fraqueza muscular, mialgia (dor muscular);
- trauma.
Raro: pode afetar 1 a 10 usuários em 10.000
- infecção;
- diminuição do número de todos os tipos de células sanguíneas;
- reações de hipersensibilidade graves (DRESS)
- diminuição da concentração de sódio no sangue;
- suicídio, transtorno de personalidade (problemas de comportamento), pensamento alterado (pensamento lento, incapacidade de concentração);
- espasmos musculares incontroláveis envolvendo cabeça, tronco e membros, dificuldade em controlar os movimentos, hipercinesia (hiperatividade);
- pancreatite;
- insuficiência hepática, hepatite;
- erupção cutânea que pode formar bolhas e aparecer como pequenos alvos (mancha escura central cercada por uma "área mais clara, com um anel escuro ao redor da borda) (eritema multiforme), uma erupção disseminada com bolhas e descamação da pele, especialmente ao redor da boca, nariz, olhos e genitais (síndrome de Stevens-Johnson) e uma forma mais grave que causa descamação da pele em mais de 30% da superfície corporal (necrólise epidérmica tóxica).
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico, incluindo quaisquer efeitos secundários possíveis não mencionados neste folheto. Você também pode relatar os efeitos colaterais diretamente através do sistema nacional de notificação. Ao relatar os efeitos colaterais, você pode ajudar a fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior após EXP: e no blister após EXP:. A data de validade refere-se ao último dia desse mês.
Este medicamento não requer quaisquer condições especiais de armazenamento.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Conteúdo da embalagem e outras informações
O que Keppra contém
O ingrediente ativo é chamado levetiracetam.
Cada comprimido de Keppra 250 mg contém 250 mg de levetiracetam.
Cada comprimido de Keppra 500 mg contém 500 mg de levetiracetam.
Cada comprimido de Keppra 750 mg contém 750 mg de levetiracetam.
Cada comprimido de Keppra 1000 mg contém 1000 mg de levetiracetam.
Os outros ingredientes são:
Núcleo do tablet: croscarmelose de sódio, macrogol 6000, sílica coloidal anidra, estearato de magnésio.
Revestimento: álcool polivinílico parcialmente hidrolisado, dióxido de titânio (E171), macrogol 3350, talco, corantes *.
* Os corantes são:
Comprimidos de 250 mg: laca de alumínio de índigo carmim (E132)
Comprimidos de 500 mg: óxido de ferro amarelo (E172)
Comprimidos de 750 mg: amarelo sol FCF (E110), óxido de ferro vermelho (E172)
Comprimidos de 1000 mg: (sem corantes adicionais).
Descrição da aparência de Keppra e conteúdo da embalagem
Os comprimidos revestidos por película de Keppra 250 mg são azuis, ovais, ranhurados e gravados com “ucb” e “250” num dos lados.
Os comprimidos revestidos por película de Keppra 500 mg são amarelos, ovais, ranhurados e gravados com “ucb” e “500” num dos lados.
Os comprimidos revestidos por película de Keppra 750 mg são cor-de-laranja, ovais, ranhurados e gravados com “ucb” e “750” num dos lados.
Os comprimidos revestidos por película de Keppra 1000 mg são brancos, ovais, ranhurados e gravados com "ucb" e "1000" num dos lados.
Os comprimidos de Keppra são acondicionados em embalagens blister colocadas em caixas de papelão contendo:
- 250 mg: 20, 30, 50, 60, 100 x 1, 100 comprimidos revestidos por película e embalagens múltiplas contendo 200 (2 embalagens de 100) comprimidos revestidos por película.
- 500 mg: 10, 20, 30, 50, 60, 100 x 1, 100, 120 comprimidos revestidos por película e embalagens múltiplas contendo 200 (2 embalagens de 100) comprimidos revestidos por película.
- 50 mg: 20, 30, 50, 60, 80, 100 x 1, 100 comprimidos revestidos por película e embalagens múltiplas contendo 200 (2 embalagens de 100) comprimidos revestidos por película.
- 1000 mg: 10, 20, 30, 50, 60, 100 x 1, 100 comprimidos revestidos por película e embalagens múltiplas contendo 200 (2 embalagens de 100) comprimidos revestidos por película.
As embalagens de 100 x 1 comprimido estão disponíveis em blisters destacáveis para dose unitária de alumínio / PVC.
Todas as outras embalagens estão disponíveis em blister padrão de alumínio / PVC.
Nem todos os tamanhos de embalagem podem ser comercializados
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
KEPPRA 500 MG COMPRIMIDOS REVESTIDOS COM PELÍCULA
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Cada comprimido revestido por película contém 500 mg de levetiracetam.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Comprimido revestido por película.
Amarelo, oval, gravado e com as palavras "ucb" e "500" gravadas numa das faces.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
Keppra é indicado como monoterapia no tratamento de crises parciais com ou sem generalização secundária em adultos e adolescentes a partir dos 16 anos de idade com epilepsia diagnosticada recentemente.
Keppra é indicado como terapia adjuvante
• no tratamento de crises parciais com ou sem generalização secundária em adultos, adolescentes, crianças e bebês a partir de 1 mês de idade com epilepsia
• no tratamento de convulsões mioclônicas em adultos e adolescentes a partir dos 12 anos de idade com epilepsia mioclônica juvenil
• no tratamento de crises tônico-clônicas generalizadas primárias em adultos e adolescentes a partir dos 12 anos de idade com epilepsia generalizada idiopática.
04.2 Posologia e método de administração
Dosagem
Monoterapia para adultos e adolescentes a partir de 16 anos
A dose inicial recomendada é 250 mg duas vezes ao dia, que deve ser aumentada para uma dose terapêutica inicial de 500 mg duas vezes ao dia após duas semanas. A dose pode ser aumentada em 250 mg duas vezes ao dia a cada duas semanas com base na resposta clínica. A dose máxima é de 1500 mg duas vezes ao dia.
Terapia complementar para adultos (≥ 18 anos) e adolescentes (12 a 17 anos) com peso igual ou superior a 50 kg
A dose terapêutica inicial é de 500 mg duas vezes ao dia. Esta dose pode ser iniciada no primeiro dia de tratamento.
Com base na resposta clínica e tolerabilidade, a dose diária pode ser aumentada até um máximo de 1500 mg duas vezes ao dia. Os ajustes posológicos podem ser feitos em aumentos ou reduções de 500 mg duas vezes ao dia a cada duas a quatro semanas.
Populações especiais
Idosos (com 65 anos ou mais)
O ajuste da dose é recomendado em pacientes idosos com insuficiência renal (ver "Insuficiência renal" abaixo).
Falência renal
A dose diária deve ser individualizada de acordo com a função renal.
Para pacientes adultos, consulte a tabela a seguir e ajuste a dosagem conforme indicado. Para usar esta tabela de dosagem, é necessário estimar a depuração da creatinina do paciente (CLcr) em ml / min. CLcr em ml / min pode ser calculado a partir da determinação da creatinina sérica (mg / dl) usando, para adultos e adolescentes com peso igual ou superior a 50 kg, a seguinte fórmula:
Além disso, CLcr é ajustado para a área de superfície corporal (BSA) da seguinte forma:
Ajuste de dosagem para pacientes adultos e adolescentes com peso superior a 50 kg e função renal comprometida:
Uma dose de carga de 750 mg é recomendada no primeiro dia de tratamento com levetiracetam.
Após a diálise, uma dose adicional entre 250 e 500 mg é recomendada.
Para crianças com insuficiência renal, a dose de levetiracetam deve ser ajustada com base na função renal, pois a depuração de levetiracetam está relacionada à função renal. Essa recomendação é baseada em um estudo realizado com pacientes adultos com insuficiência renal.
Em adolescentes jovens, crianças e bebês, CLcr, em ml / min / 1,73 m2, pode ser estimado a partir da determinação da creatinina sérica (em mg / dl) usando a seguinte fórmula (fórmula de Schwartz):
ks = 0,45 em bebês nascidos a termo com idade até 1 ano; ks = 0,55 em crianças menores de 13 anos e em adolescentes do sexo feminino; ks = 0,7 em adolescentes do sexo masculino.
Ajuste de dosagem para bebês, crianças e adolescentes com peso inferior a 50 kg com insuficiência renal:
Keppra solução oral deve ser usado em doses abaixo de 250 mg e em pacientes que não conseguem engolir os comprimidos.
Uma dose de carga de 10,5 mg / kg (0,105 ml / kg) é recomendada no primeiro dia de tratamento com levetiracetam.
Uma dose de carga de 15 mg / kg (0,15 ml / kg) é recomendada no primeiro dia de tratamento com levetiracetam.
Após a diálise, uma dose suplementar de 3,5 a 7 mg / kg (0,035 a 0,07 ml / kg) é recomendada.
Após a diálise, é recomendada uma dose adicional de 5 a 10 mg / kg (0,05 a 0,10 ml / kg).
Insuficiência Hepática
Não é necessário ajuste da dose em pacientes com insuficiência hepática leve a moderada. Em pacientes com insuficiência hepática grave, a depuração da creatinina pode subestimar o grau de insuficiência renal. Portanto, uma redução de 50% da dose de manutenção diária é recomendada quando a depuração da creatinina é 2.
População pediátrica
O médico deve prescrever a forma farmacêutica e a dosagem mais adequadas com base na idade, peso e dose.
A formulação em comprimido não é adequada para uso em bebês e crianças com menos de 6 anos de idade.Keppra solução oral é a formulação preferida para uso nesta população. Além disso, as dosagens disponíveis dos comprimidos não são adequadas para o tratamento inicial em crianças com peso inferior a 25 kg, para doentes que não conseguem engolir os comprimidos ou para administrar doses inferiores a 250 mg. Em todos os casos mencionados acima, Keppra solução oral deve ser usado.
Monoterapia
A segurança e eficácia de Keppra administrado em monoterapia a crianças e adolescentes com menos de 16 anos não foram estabelecidas.
Não há dados disponíveis.
Terapia adjuvante para bebês de 6 a 23 meses de idade, crianças (2 a 11 anos) e adolescentes (12 a 17 anos) com peso inferior a 50 kg
Keppra solução oral é a formulação preferida para uso em bebês e crianças com idade inferior a 6 anos.
A dose terapêutica inicial é de 10 mg / kg duas vezes ao dia.
Com base na resposta clínica e tolerabilidade, a dose pode ser aumentada até 30 mg / kg duas vezes ao dia. Os ajustes posológicos não devem exceder aumentos ou diminuições de 10 mg / kg duas vezes ao dia a cada duas semanas. Deve ser usada a menor dose eficaz.
A dose em crianças com 50 kg ou mais é a mesma que em adultos.
Dose recomendada para bebês a partir dos 6 meses de idade, crianças e adolescentes:
Crianças com peso igual ou inferior a 25 kg devem iniciar preferencialmente o tratamento com Keppra 100 mg / ml solução oral.
A dose em crianças e adolescentes com peso igual ou superior a 50 kg é igual à dos adultos.
Terapia complementar para bebês de 1 mês a menos de 6 meses de idade
A solução oral é a formulação para uso em bebês.
Método de administração
Os comprimidos revestidos por película devem ser administrados por via oral, engolidos com uma quantidade suficiente de líquido e podem ser tomados com ou sem alimentos. A dose diária deve ser dividida ao meio em duas administrações.
04.3 Contra-indicações
Hipersensibilidade à substância ativa ou a outros derivados da pirrolidona ou a qualquer um dos excipientes mencionados na secção 6.1.
04.4 Advertências especiais e precauções adequadas de uso
Descontinuação do tratamento
De acordo com a prática clínica atual, uma retirada gradual é recomendada se o tratamento com Keppra for descontinuado (por exemplo, em adultos e adolescentes com peso superior a 50 kg: diminuir em 500 mg duas vezes ao dia em intervalos incluindo entre duas e quatro semanas; em bebês acima 6 meses de idade, em crianças e adolescentes com peso inferior a 50 kg: a redução da dose não deve exceder 10 mg / kg duas vezes ao dia a cada duas semanas; em bebês (menos de 6 meses de idade): a redução da dose não deve exceder 7 mg / kg duas vezes por dia a cada duas semanas).
Falência renal
A administração de Keppra a pacientes com insuficiência renal pode exigir ajuste da dose. Em doentes com compromisso hepático grave, recomenda-se avaliar a função renal antes de estabelecer a posologia (ver secção 4.2).
Suicídio
Foram notificados casos de suicídio, tentativa de suicídio, ideação suicida e comportamento suicida em doentes tratados com antiepilépticos (incluindo levetiracetam). Uma meta-análise de ensaios clínicos randomizados e controlados por placebo com medicamentos antiepilépticos mostrou um risco ligeiramente aumentado de ideação e comportamento suicida. O mecanismo desse risco não é conhecido.
Consequentemente, os pacientes devem ser monitorados quanto a sinais de depressão e / ou ideação e comportamento suicida, e o tratamento apropriado deve ser considerado. Os pacientes (e cuidadores) devem ser informados de que, se surgirem sinais de depressão e / ou ideação ou comportamento suicida, deve-se procurar atendimento médico.
População pediátrica
A formulação do comprimido não é adequada para uso em bebês e crianças menores de 6 anos.
Os dados disponíveis em crianças não sugerem uma influência no crescimento e na puberdade. No entanto, os efeitos de longo prazo na aprendizagem, inteligência, crescimento, função endócrina, puberdade e potencial reprodutivo em crianças são desconhecidos.
A segurança e eficácia do levetiracetam não foram totalmente avaliadas em crianças com idade inferior a 1 ano com epilepsia. Em estudos clínicos, apenas 35 crianças com idade <1 ano com crises parciais foram expostas a Keppra, das quais apenas 13 tinham idade inferior a 6 meses.
04.5 Interações com outros medicamentos e outras formas de interação
Medicamentos antiepilépticos
Os dados de estudos clínicos pré-comercialização realizados em adultos indicam que Keppra não afeta as concentrações séricas dos antiepilépticos existentes (fenitoína, carbamazepina, ácido valpróico, fenobarbital, lamotrigina, gabapentina e primidona) e que estes antiepilépticos não afetam a farmacocinética de Keppra.
Tal como em adultos, em doentes pediátricos administrados com doses de levetiracetam até 60 mg / kg / dia, não há evidência de interações clinicamente significativas com outros medicamentos.
Uma avaliação retrospectiva das interações farmacocinéticas em crianças e adolescentes com epilepsia (4 a 17 anos) confirmou que a terapia adjuvante com levetiracetam administrado por via oral não afetou as concentrações séricas no estado estacionário de carbamazepina e valproato administrados concomitantemente. No entanto, os dados sugeriram uma depuração de levetiracetam 20% mais elevada em crianças a tomar medicamentos antiepilépticos indutores de enzimas. Nenhum ajuste de dose é necessário.
Probenecida
Probenecida (500 mg quatro vezes ao dia), um agente bloqueador da secreção tubular renal, demonstrou inibir a depuração renal do metabólito primário, mas não do levetiracetam. No entanto, a concentração desse metabólito permanece baixa. Prevê-se que outros medicamentos excretados com secreção tubular ativa reduzam a depuração renal do metabolito. O efeito do levetiracetam sobre a probenecida não foi estudado e o efeito do levetiracetam sobre outros medicamentos secretados ativamente, por exemplo. AINEs, sulfonamidas e metotrexato, é desconhecido.
Contraceptivos orais e outras interações farmacocinéticas
Levetiracetam 1000 mg por dia não afetou a farmacocinética dos contraceptivos orais (etinilestradiol e levonorgestrel); os parâmetros endócrinos (hormônio luteinizante e progesterona) não foram modificados. Levetiracetam 2000 mg por dia não afetou a farmacocinética da digoxina e varfarina; os tempos de protrombina não foram alterados. A administração concomitante de digoxina, contraceptivos orais e varfarina não afetou a farmacocinética do levetiracetam.
Antiácidos
Não existem dados disponíveis sobre a influência dos antiácidos na absorção do levetiracetam.
Laxantes
Têm havido notificações isoladas de diminuição da eficácia do levetiracetam quando o laxante osmótico macrogol foi administrado concomitantemente com levetiracetam oral. Portanto, o macrogol não deve ser tomado por via oral entre uma hora antes e uma hora depois de tomar levetiracetam.
Comida e álcool
A extensão da absorção do levetiracetam não foi afetada pelos alimentos, mas a taxa de absorção foi ligeiramente reduzida.
Não existem dados sobre as interações do levetiracetam com o álcool.
04.6 Gravidez e lactação
Gravidez
Os dados pós-comercialização de vários registros de gravidez prospectiva documentaram os resultados da exposição à monoterapia com levetiracetam em mais de 1000 mulheres durante o primeiro trimestre da gravidez. No geral, esses dados não sugerem um aumento substancial no risco de malformações congênitas maiores, embora um risco teratogênico não possa ser completamente excluído. A terapia com vários AEDs está associada a um risco maior de malformações congênitas do que a monoterapia e, portanto, a monoterapia deve ser considerada. Os estudos em animais revelaram toxicidade reprodutiva (ver secção 5.3).
Keppra não é recomendado, a menos que seja clinicamente necessário, durante a gravidez e em mulheres com potencial para engravidar que não estejam a utilizar métodos contraceptivos.
Tal como acontece com outros medicamentos antiepilépticos, as alterações fisiológicas associadas à gravidez podem afetar as concentrações plasmáticas de levetiracetam. Durante a gravidez, foram observadas concentrações plasmáticas diminuídas de levetiracetam. Esta redução é mais pronunciada durante o terceiro trimestre (até 60% da concentração basal antes da gravidez). Mulheres grávidas tratadas com levetiracetam devem ser cuidadosamente acompanhadas do ponto de vista clínico. A descontinuação dos tratamentos antiepilépticos pode levar a uma exacerbação da doença, que pode ser prejudicial à mãe e ao feto.
Hora da alimentação
O levetiracetam é excretado no leite materno humano. Portanto, a amamentação não é recomendada, porém, se o tratamento com levetiracetam se tornar necessário durante a amamentação, a relação benefício / risco do tratamento deve ser avaliada, levando em consideração a importância da amamentação.
Fertilidade
Não foi encontrado impacto na fertilidade em estudos com animais (ver secção 5.3). Não há dados clínicos disponíveis; o risco potencial em humanos é desconhecido.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
Não foram realizados estudos sobre a capacidade de conduzir e utilizar máquinas.
Dada a possível sensibilidade individual diferente, alguns pacientes podem sentir sonolência ou outros sintomas relacionados com a ação no sistema nervoso central, especialmente no início do tratamento ou após um aumento da dose. Portanto, recomenda-se cautela em pacientes que estão envolvidos em atividades que requerem alta concentração, como dirigir veículos ou operar máquinas. Os pacientes devem ser aconselhados a não dirigir ou operar máquinas até que seja estabelecido que sua capacidade de realizar essas atividades não foi afetada.
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
O perfil de eventos adversos apresentado abaixo é baseado na análise de ensaios clínicos controlados por placebo agrupados em todas as indicações estudadas para um total de 3416 pacientes tratados com levetiracetam. Estes dados são complementados com o uso de levetiracetam em estudos de extensão abertos correspondentes, também As reações adversas notificadas com mais frequência foram nasofaringite, sonolência, cefaleia, fadiga e tonturas. O perfil de segurança do levetiracetam é geralmente semelhante em todos os grupos etários (doentes adultos e pediátricos) e as indicações aprovadas para o tratamento de epilepsia.
Tabela de reações adversas
As reações adversas notificadas em ensaios clínicos (adultos, adolescentes, crianças e bebés com mais de 1 mês de idade) e na experiência pós-comercialização estão listadas na seguinte tabela por classes de sistemas de órgãos e frequência. É definida como se segue: muito frequentes (≥1 / 10); comum (≥1 / 100,
Descrição das reações adversas selecionadas
O risco de anorexia é maior quando o topiramato é coadministrado com levetiracetam.
Em vários casos de alopecia, a cura foi observada após a interrupção do tratamento com levetiracetam.
A supressão da medula óssea foi identificada em alguns casos de pancitopenia.
População pediátrica
Em doentes com idades entre 1 mês e menos de 4 anos, um total de 190 doentes foram tratados com levetiracetam em estudos de extensão controlados por placebo e abertos. Sessenta desses pacientes foram tratados com levetiracetam em estudos controlados com placebo. Em pacientes com idade entre 4 e 16 anos, um total de 645 pacientes foram tratados com levetiracetam em estudos de extensão controlados por placebo e abertos. 233 desses pacientes foram tratados com levetiracetam em estudos controlados com placebo. Em ambas as faixas etárias pediátricas, esses dados são integrados à experiência pós-comercialização com o uso de levetiracetam.
O perfil de eventos adversos do levetiracetam é geralmente semelhante em todos os grupos etários e nas indicações aprovadas para a epilepsia. Em estudos clínicos controlados com placebo, os resultados de segurança em pacientes pediátricos foram consistentes com o perfil de segurança de levetiracetam em adultos, com exceção de reações adversas comportamentais e psiquiátricas que foram mais comuns em crianças do que em adultos. Em crianças e adolescentes de 4 a 16 anos, vômitos (muito comuns, 11,2%), agitação (comum, 3,4%) foram relatados com mais frequência do que em outras faixas etárias ou no perfil de segurança geral.), Alterações de humor (comuns, 2,1 %), labilidade afetiva (comum, 1,7%), agressão (comum, 8,2%), comportamento anormal (comum, 5,6%) e letargia (comum, 3,9%) Em bebês e crianças de 1 mês a menos de 4 anos, irritabilidade foi relatada com mais frequência do que em outras faixas etárias ou no perfil de segurança geral (muito comum, 11,7%) e coordenação anormal (comum, 3,3%).
Um estudo de segurança em pacientes pediátricos, conduzido de acordo com um desenho de não inferioridade, duplo-cego, controlado por placebo, avaliou os efeitos cognitivos e neuropsicológicos de Keppra em crianças de 4 a 16 anos de idade com crises parciais. Keppra não demonstrou ser diferente (não inferior) do placebo na alteração da linha de base na pontuação obtida no subteste "Atenção e Memória" da escala Leiter-R (Pontuação composta da tela de memória) na população por protocolo. Os resultados relacionados às funções comportamentais e emocionais indicaram uma piora, nos pacientes tratados com Keppra, do comportamento agressivo medido de forma padronizada e sistemática, com o uso de uma ferramenta validada (CBCL - Lista de verificação de comportamento infantil de Achenbach) No entanto, os indivíduos que tomaram Keppra no estudo aberto de acompanhamento de longo prazo não experimentaram, em média, deterioração em suas funções comportamentais e emocionais; em particular, as avaliações de agressão em comportamentos não se deterioraram em comparação com a linha de base.
04.9 Overdose
Sintomas
Foram observados sonolência, agitação, agressão, diminuição do nível de consciência, depressão respiratória e coma com sobredosagens de Keppra.
Tratamento de overdose
Após uma overdose aguda, o estômago pode ser esvaziado por lavagem gástrica ou indução de vômito. Não existe um antídoto específico para o levetiracetam. O tratamento da sobredosagem com levetiracetam deve ser sintomático e pode incluir hemodiálise.A eficácia da extração por diálise é de 60% para o levetiracetam e 74% para o metabolito primário.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo Farmacoterapêutico: antiepilépticos, outros antiepilépticos, código ATC: N03AX14.
A substância ativa, o levetiracetam, é um derivado da pirrolidona (enantiómero S de α-etil-2-oxo-1-pirrolidina acetamida), quimicamente não relacionado com substâncias antiepilépticas existentes.
Mecanismo de ação
O mecanismo de ação do levetiracetam ainda não foi totalmente explicado, mas parece ser diferente dos mecanismos dos medicamentos antiepilépticos atuais. em vitro e na Vivo sugerem que o levetiracetam não altera as características celulares básicas e a neurotransmissão normal.
Educação em vitro mostram que o levetiracetam atua nos níveis intraneuronais de Ca2 + inibindo parcialmente as correntes de Ca2 + do tipo N e reduzindo a liberação de Ca2 + dos locais de armazenamento intraneuronal. Além disso, reverte parcialmente a redução, induzida por zinco e β-carbolina, das correntes induzidas por GABA e glicina. Educação em vitro eles também descobriram que o levetiracetam se liga a um local específico no tecido cerebral de roedores. Este local de ligação é a proteína 2A da vesícula sináptica, que se acredita estar envolvida na fusão da vesícula e na exocitose do neurotransmissor. O levetiracetam e análogos relacionados mostram um grau de afinidade para a ligação à proteína 2A da vesícula sináptica que se correlaciona com a potência de sua proteção antiepiléptica no sistema audiogênico modelo de epilepsia em camundongos. Esse achado sugere que a interação entre o levetiracetam e a proteína 2A da vesícula sináptica parece desempenhar um papel no mecanismo de ação antiepiléptica do fármaco.
Efeitos farmacodinâmicos
O levetiracetam induz ação protetora em um amplo espectro de modelos animais de epilepsia parcial e primária generalizada, sem efeito pró-convulsivo, sendo o metabólito primário inativo.
Em humanos, a atividade em condições de epilepsia parcial e generalizada (descarga epiléptica / resposta fotoparoxística) confirmou o amplo espectro do perfil farmacológico do levetiracetam.
Eficácia clínica e segurança
Terapia adjuvante no tratamento de crises parciais com ou sem generalização secundária em adultos, adolescentes, crianças e bebês a partir de 1 mês de idade com epilepsia.
Em adultos, a eficácia do levetiracetam foi demonstrada em 3 estudos duplo-cegos, controlados por placebo, com doses de 1000 mg, 2000 mg ou 3000 mg / dia, divididas em 2 doses, para uma duração de tratamento de até 18 semanas. análise a porcentagem de pacientes que alcançaram uma redução na frequência de crises de início parcial por semana, no período de tratamento de dose estável (12/14 semanas), igual ou superior a 50% da linha de base, foi 27, 7%, 31,6% e 41,3 % dos pacientes tratados com 1.000, 2.000 ou 3.000 mg de levetiracetam, respectivamente, e 12,6% para os pacientes tratados com placebo.
População pediátrica
A eficácia do levetiracetam em pacientes pediátricos (4 a 16 anos de idade) foi demonstrada em um estudo duplo-cego, controlado por placebo, que incluiu 198 pacientes e teve uma duração de tratamento de 14 semanas. Neste estudo, os pacientes receberam levetiracetam em regime fixo dose de 60 mg / kg / dia (duas vezes ao dia).
44,6% dos pacientes tratados com levetiracetam e 19,6% dos pacientes tratados com placebo tiveram uma redução de 50% ou mais na frequência de crises parciais por semana desde o início. Com a continuação do tratamento de longo prazo, 11,4% dos pacientes ficaram sem crises por pelo menos 6 meses e 7,2% não tiveram crises por pelo menos 1 ano.
Em pacientes pediátricos (1 mês a menos de 4 anos de idade), a eficácia do levetiracetam foi demonstrada em um estudo duplo-cego, controlado por placebo, que incluiu 116 pacientes e teve uma duração de tratamento de 5 dias. foram prescritos uma dose diária de 20 mg / kg, 25 mg / kg, 40 mg / kg ou 50 mg / kg de solução oral com base em seu esquema de titulação de dose relacionado à idade. as seguintes doses foram usadas: 20 mg / kg / dia , titulado para 40 mg / kg / dia, para bebês de um mês a menos de seis meses de idade; 25 mg / kg / dia, titulado para 50 mg / kg / dia para bebês e crianças de 6 meses a menos de 4 anos de idade A dose diária total foi dividida em duas administrações por dia.
A principal medida da eficácia do tratamento foi a taxa de resposta dos pacientes (porcentagem de pacientes com ≥50% de redução na frequência média diária de crises parciais na linha de base), avaliada por um único examinador cego usando vídeo EEG por um período de 48 horas. A análise de eficácia foi realizada em 109 pacientes submetidos a vídeo EEG por pelo menos 24 horas, tanto durante o período inicial quanto durante o período de avaliação. 43,6% dos pacientes tratados com Levetiracetam e 19,6% dos pacientes tratados com placebo foram considerados responsivos. Os resultados foram consistentes em todos os grupos etários. No tratamento contínuo a longo prazo, 8,6% dos pacientes ficaram sem crises por pelo menos 6 meses e 7,8% não tiveram crises por pelo menos 1 ano.
Monoterapia no tratamento de crises parciais com ou sem generalização secundária em pacientes a partir dos 16 anos de idade com epilepsia recém-diagnosticada.
A eficácia da monoterapia com levetiracetam foi demonstrada em um estudo de não inferioridade comparativo de grupo paralelo duplo-cego versus carbamazepina (CR) de liberação controlada em 576 pacientes com 16 anos ou mais com epilepsia nova ou nova. Recentemente diagnosticado. Os pacientes foram obrigados a têm apenas convulsões parciais não provocadas ou convulsões tônico-clônicas generalizadas. Os pacientes foram randomizados para carbamazepina CR 400 - 1200 mg / dia ou levetiracetam 1000 - 3000 mg / dia e o tratamento durou até 121 semanas com base na resposta.
A liberdade das crises por um período de 6 meses foi alcançada em 73,0% dos pacientes tratados com levetiracetam e em 72,8% dos pacientes tratados com carbamazepina CR; a diferença absoluta corrigida entre os tratamentos foi de 0,2% (IC 95%: 7,8 - 8,2). Mais da metade dos indivíduos permaneceu sem crises por 12 meses (56,6% e 58,5% dos indivíduos tratados com levetiracetam e carbamazepina CR, respectivamente).
Num estudo que reflecte a prática clínica, o tratamento antiepiléptico concomitante pode ser suspenso num número limitado de doentes que responderam à terapêutica adjuvante com levetiracetam (36 de 69 doentes adultos).
Terapia adjuvante no tratamento das crises mioclônicas em adultos e adolescentes a partir dos 12 anos de idade com epilepsia mioclônica juvenil.
A eficácia do levetiracetam foi demonstrada em um estudo duplo-cego, controlado por placebo de 16 semanas em pacientes com 12 anos ou mais com epilepsia generalizada idiopática com convulsões mioclônicas em diferentes síndromes. A maioria dos pacientes tinha epilepsia mioclônica juvenil.
Neste estudo, a dose de levetiracetam foi de 3.000 mg / dia administrada em duas doses divididas.
58,3% dos pacientes tratados com levetiracetam e 23,3% dos pacientes tratados com placebo tiveram pelo menos uma redução de 50% nos dias de convulsão mioclônica por semana. Após tratamento contínuo de longo prazo, 28,6% dos pacientes ficaram livres de crises mioclônicas por pelo menos 6 meses e 21,0% dos pacientes ficaram livres de crises mioclônicas por pelo menos 1 ano.
Terapia adjuvante no tratamento de crises tônico-clônicas generalizadas primárias em adultos e adolescentes a partir dos 12 anos de idade com epilepsia generalizada idiopática.
A eficácia do levetiracetam foi demonstrada em um estudo duplo-cego, controlado por placebo de 24 semanas que incluiu adultos, adolescentes e um número limitado de crianças com epilepsia generalizada idiopática com crises tônico-clônicas generalizadas primárias (PGTCs). Em diferentes síndromes (juvenil epilepsia mioclônica, epilepsia ausência juvenil, epilepsia ausência infantil ou epilepsia com convulsão do Grande Macho ao despertar). Neste estudo, a dose de levetiracetam foi de 3000 mg / dia para adultos e adolescentes ou 60 mg / kg / dia para crianças, administrada em duas doses divididas.
72,2% dos pacientes tratados com levetiracetam e 45,2% dos pacientes tratados com placebo tiveram uma redução de 50% ou mais na frequência de convulsões de PGTC por semana. Após a continuação do tratamento de longo prazo, 47,4% dos pacientes estavam livres de crises tônico-clônicas por pelo menos 6 meses e 31,5% estavam livres de crises tônico-clônicas por pelo menos 1 ano.
05.2 Propriedades farmacocinéticas
O levetiracetam é um composto altamente solúvel e permeável. O perfil farmacocinético é linear com pouca variabilidade intra e interindividual. Não há alteração na depuração após administração repetida.Não há evidência de qualquer variação circadiana relevante e de gênero e raça. O perfil farmacocinético é comparável em voluntários saudáveis e em pacientes com epilepsia.
Dada a sua absorção completa e linear, os níveis plasmáticos de levetiracetam podem ser previstos a partir da dose oral expressa em mg / kg de peso corporal. Portanto, não há necessidade de monitorar os níveis plasmáticos de levetiracetam.
Houve uma correlação significativa entre a saliva e as concentrações plasmáticas em adultos e crianças (a proporção de saliva / concentrações plasmáticas variou de 1 a 1,7 para a formulação de comprimido oral e, após 4 horas da "ingestão, para a formulação de solução oral).
Adultos e adolescentes
Absorção
O levetiracetam é rapidamente absorvido após administração oral. A biodisponibilidade oral é próxima a 100%.
As concentrações plasmáticas máximas (Cmax) são atingidas 1,3 horas após a administração.O estado estacionário é alcançado após dois dias de duas doses diárias.
As concentrações plasmáticas máximas (Cmax) são tipicamente 31 e 43 mcg / mL após uma dose única de 1000 mg e uma dose repetida de 1000 mg duas vezes ao dia, respectivamente.
A extensão da absorção não depende da dose e não é afetada pelos alimentos.
Distribuição
Não existem dados sobre a distribuição nos tecidos em humanos.
Nem o levetiracetam nem seu metabólito primário se ligam significativamente às proteínas plasmáticas (
O volume de distribuição do levetiracetam é de aproximadamente 0,5 a 0,7 l / kg e está próximo do volume corporal total de água.
Biotransformação
O levetiracetam não é extensivamente metabolizado em humanos.A principal via metabólica (24% da dose) é a hidrólise enzimática do grupo da acetamida. A produção do metabólito primário, ucb L057, não é suportada pelas isoformas do citocromo P450 hepático. A hidrólise do grupo acetamida foi mensurável em vários tecidos, incluindo células sanguíneas.O metabólito ucb L057 é farmacologicamente inativo.
Dois metabólitos menores também foram identificados. Um foi obtido da hidroxilação do anel da pirrolidona (1,6% da dose) e o outro da abertura do anel da pirrolidona (0,9% da dose).
Outros componentes desconhecidos representaram apenas 0,6% da dose.
Na Vivo não houve evidência de interconversão enantiomérica para levetiracetam ou seu metabólito primário.
Em vitro, o levetiracetam e seu metabólito primário demonstraram não inibir as atividades das principais isoformas do citocromo P450 hepático humano (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 e 1A2), glucuronil transferase (UGT1A1 e UGT1A6 além de hidróxido de epóxido) , o levetiracetam não afeta a glucuronidação em vitro de ácido valpróico.
Em culturas de hepatócitos humanos, o levetiracetam teve pouco ou nenhum efeito no CYP1A2, SULT1E1 ou UGT1A1. O levetiracetam causou indução moderada do CYP2B6 e CYP3A4. Os dados em vitro e os dados na Vivo relacionados à interação com contraceptivos orais, digoxina e varfarina, indicam que nenhuma indução enzimática significativa é esperada na Vivo. Conseqüentemente, a interação de Keppra com outras substâncias, ou o contrário, É improvável.
Eliminação
A meia-vida plasmática em adultos é de 7 ± 1 horas e não muda com a dose, via de administração ou administração repetida.A depuração corporal total média é de 0,96 ml / min / kg.
A principal via de excreção é a via urinária, responsável em média pela eliminação de 95% da dose administrada (aproximadamente 93% da dose é excretada em 48 horas) .A eliminação fecal é responsável por apenas 0,3% da dose.
A excreção urinária cumulativa do levetiracetam e do seu metabólito primário é responsável pela eliminação de 66% e 24% da dose, respectivamente, nas primeiras 48 horas.
A depuração renal de levetiracetam e ucb L057 é de 0,6 e 4,2 ml / min / kg, respectivamente, indicando que o levetiracetam é excretado por filtração glomerular com subsequente reabsorção tubular e que o metabólito primário também é excretado por secreção tubular ativa além da filtração glomerular. A eliminação do levetiracetam está relacionada com a depuração da creatinina.
Cidadãos idosos
Nos "idosos", a meia-vida aumentou cerca de 40% (10 a 11 horas). Isto deve-se à diminuição da função renal nesta população (ver secção 4.2).
Falência renal
A depuração corporal aparente do levetiracetam e de seu metabólito primário correlaciona-se com a depuração da creatinina. Por conseguinte, é recomendado ajustar a dose diária de manutenção de Keppra, com base na depuração da creatinina em doentes com compromisso renal moderado e grave (ver secção 4.2).
Em indivíduos adultos com doença renal anúrica terminal, a meia-vida foi de aproximadamente 25 e 3,1 horas nos períodos de inter-diálise e durante os períodos de diálise, respectivamente.
A fração de levetiracetam removida foi de 51% durante uma diálise típica de 4 horas.
Insuficiência Hepática
Em indivíduos com insuficiência hepática leve e moderada, nenhuma modificação significativa da depuração do levetiracetam foi encontrada. Na maioria dos indivíduos com insuficiência hepática grave, a depuração do levetiracetam foi reduzida em mais de 50% devido à insuficiência renal concomitante (ver secção 4.2).
População pediátrica
Crianças (de 4 a 12 anos)
Após uma administração oral única (20 mg / kg) em crianças (6 a 12 anos) com epilepsia, a meia-vida do levetiracetam foi de 6,0 horas.A depuração corrigida do peso corporal aparente foi aproximadamente 30% maior do que em adultos com epilepsia.
Após administração oral de dose repetida (20 a 60 mg / kg / dia) a crianças epilépticas (4 a 12 anos), o levetiracetam foi rapidamente absorvido. As concentrações plasmáticas máximas foram observadas 0,5 a 1,0 horas após a administração. Observaram-se aumentos lineares e proporcionais à dose para as concentrações plasmáticas máximas e área sob a curva.A meia-vida de eliminação foi de aproximadamente 5 horas. A depuração corporal aparente foi de 1,1 mL / min / kg.
Bebês e crianças (1 mês a 4 anos)
Após a administração de uma dose única (20 mg / kg) de solução oral de 100 mg / ml a crianças epilépticas (1 mês a 4 anos), o levetiracetam foi rapidamente absorvido e foram observados picos de concentração plasmática aproximadamente 1 hora após a administração. Os resultados farmacocinéticos indicaram que a meia-vida é mais curta (5,3 horas) do que em adultos (7,2 horas) e a depuração aparente foi mais rápida (1,5 ml / min / kg) do que em adultos (0, 96 ml / min / kg).
Em análises farmacocinéticas populacionais conduzidas em pacientes de 1 mês a 16 anos de idade, o peso corporal foi significativamente correlacionado com a depuração aparente (depuração aumentou com o aumento do peso corporal) e o volume aparente de distribuição.A idade também afetou ambos os parâmetros. Esse efeito foi acentuado para os bebês mais jovens e atenuado com o aumento da idade, tornando-se insignificante por volta dos 4 anos de idade.
Em ambas as análises farmacocinéticas populacionais, houve um aumento de aproximadamente 20% na depuração aparente do levetiracetam quando coadministrado com um fármaco antiepiléptico indutor de enzimas.
05.3 Dados de segurança pré-clínica
Os dados não clínicos não revelam risco para o ser humano, segundo estudos convencionais de farmacologia de segurança, genotoxicidade e potencial carcinogénico.
Os eventos adversos não observados em estudos clínicos, mas observados em ratos e em menor extensão em camundongos, em níveis de exposição semelhantes aos níveis de exposição humana e com possível relevância para uso clínico, foram os índices de resposta às alterações hepáticas. Adaptativos, como ganho de peso e hipertrofia centrolobular, infiltração adiposa e elevação das enzimas hepáticas no plasma.
Nenhum efeito adverso na fertilidade masculina e feminina ou capacidade reprodutiva foi observado em ratos com doses de até 1800 mg / kg / dia (6 vezes o MRHD (Dose Diária Humana Máxima Recomendada) com base em mg / m2 ou com base na exposição), tanto na geração parental quanto na geração F1.
Dois estudos de desenvolvimento embriofetal (EFD: Desenvolvimento Embrião-Fetal) foram conduzidos em ratos a 400, 1200 e 3600 mg / kg / dia. Com 3600 mg / kg / dia, em apenas um dos 2 estudos EFD, houve uma ligeira diminuição no peso fetal associada a um aumento marginal nas alterações esqueléticas / anomalias menores. Não houve efeito na mortalidade embrionária nem aumento na incidência de malformações.Nenhum nível de efeito adverso observado) foi de 3600 mg / kg / dia para ratas grávidas (12 vezes a dose diária humana máxima recomendada (MRHD) com base em mg / m2) e 1200 mg / kg / dia para fetos.
Quatro estudos de desenvolvimento embriofetal foram conduzidos em coelhos usando doses de 200, 600, 800, 1200 e 1800 mg / kg / dia. A dose de 1800 mg / kg / dia induziu marcada toxicidade materna e diminuição do peso fetal em associação com uma maior incidência de fetos com anomalias cardiovasculares / esqueléticas. O NOAEL foi 2).
Um estudo de desenvolvimento peri e pós-natal foi conduzido em ratos com doses de levetiracetam de 70, 350, 1800 mg / kg / dia. O NOAEL foi ≥ 1800 mg / kg / dia para mulheres F0 e para a geração F1 para sobrevivência, crescimento e desenvolvimento até o desmame (6 vezes o MRHD em uma base de mg / m2).
Estudos em ratos e cães em animais recém-nascidos e jovens demonstraram que nenhum efeito adverso ocorre em nenhum dos pontos finais de desenvolvimento ou maturação padrão com doses de até 1800 mg / kg / dia (6-17 vezes o MRHD com base em mg / m2).
Avaliação de risco ambiental (Avaliação de Risco Ambiental, ERA)
É improvável que a utilização de Keppra de acordo com a informação do resumo das características do produto resulte num impacto ambiental inaceitável (ver secção 6.6).
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Núcleo:
Croscarmelose de sódio
Macrogol 6000
Sílica coloidal anidra
Estearato de magnesio
Revestimento Opadry 85F32004:
Álcool polivinílico parcialmente hidrolisado
Dióxido de titânio (E171)
Macrogol 3350
Talco
Óxido de ferro amarelo (E172)
06.2 Incompatibilidade
Não é relevante.
06.3 Período de validade
3 anos.
06.4 Precauções especiais de armazenamento
Este medicamento não requer quaisquer condições especiais de armazenamento.
06.5 Natureza da embalagem primária e conteúdo da embalagem
Blisters de alumínio / PVC colocados em caixas de papelão contendo 10, 20, 30, 50, 60, 100, 120 comprimidos revestidos por película e embalagens múltiplas contendo 200 (2 embalagens de 100) comprimidos revestidos por película.
Blisters de dose unitária perfurados de alumínio / PVC colocados em caixas de cartão contendo 100 x 1 comprimidos revestidos por película.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
UCB Pharma SA
Allée de la Recherche 60
B - 1070 Bruxelas
Bélgica
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU / 1/00/146/006 - AIC 035039066
EU / 1/00/146/007 - AIC 035039078
EU / 1/00/146/008 - AIC 035039080
EU / 1/00/146/009 - AIC 035039092
EU / 1/00/146/010 - AIC 035039104
EU / 1/00/146/011 - AIC 035039116
EU / 1/00/146/012 - AIC 035039128
EU / 1/00/146/013 - AIC 035039130
EU / 1/00/146/035 - AIC 035039332
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 29 de setembro de 2000
Data da renovação mais recente: 29 de setembro de 2010
10.0 DATA DE REVISÃO DO TEXTO
agosto de 2013