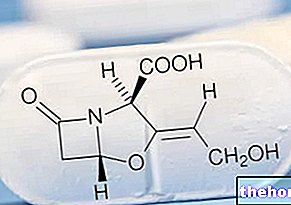
Ingredientes ativos: fumarato de dimetila (fumaras de dimetila)
Tecfidera 120 mg cápsulas gastrorresistentes
Tecfidera 240 mg cápsulas gastrorresistentes
Indicações Por que é usado o Tecfidera? Para que serve?
O que é Tecfidera
O Tecfidera é um medicamento que contém a substância ativa fumarato de dimetilo.
O que é Tecfidera
Tecfidera é usado para tratar a esclerose múltipla (EM) recorrente-remitente.
A esclerose múltipla (EM) é uma doença crônica que afeta o sistema nervoso central (SNC), incluindo o cérebro e a medula espinhal. A EM recorrente-remitente é caracterizada por ataques repetidos (recaídas) de sintomas que afetam o sistema nervoso. Os sintomas variam de paciente para paciente, mas geralmente incluem dificuldade para andar, sensação de desequilíbrio e dificuldade de visão. Esses sintomas podem desaparecer completamente quando a recidiva for resolvida, mas alguns problemas podem permanecer.
Como funciona o Tecfidera
Tecfidera parece funcionar evitando que o sistema de defesa do corpo danifique o cérebro e a medula espinhal. Isso também pode ajudar a retardar o agravamento futuro da esclerose múltipla.
Contra-indicações Quando Tecfidera não deve ser usado
Não tome Tecfidera:
- se você é alérgico ao fumarato de dimetila ou a qualquer outro ingrediente deste medicamento
Precauções de uso O que você precisa saber antes de tomar Tecfidera
Tecfidera pode afetar o número de glóbulos brancos no sangue, rins e fígado. Antes de começar a tomar Tecfidera, o seu médico fará uma análise ao sangue para contar o número dos seus glóbulos brancos e verificar se os seus rins e fígado estão a funcionar bem. O médico realizará exames periodicamente durante o tratamento. Se a contagem dos seus glóbulos brancos cair durante o tratamento, o seu médico pode considerar interromper o tratamento.
Fale com o seu médico antes de tomar Tecfidera se tiver:
- doença renal grave
- doença hepática grave
- doença do estômago ou intestino
- uma "infecção grave (como pneumonia)
Crianças e adolescentes
Tecfidera não deve ser utilizado em crianças e adolescentes com menos de 18 anos. A segurança e eficácia de Tecfidera não são conhecidas neste grupo etário.
Interações Quais medicamentos ou alimentos podem alterar o efeito de Tecfidera
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos, especialmente:
- Medicamentos que contêm ésteres de ácido fumárico (fumaratos) usados para tratar a psoríase.
- Medicamentos que afetam o sistema imunológico do corpo, incluindo outros medicamentos usados para tratar a esclerose múltipla, como fingolimode, natalizumabe ou mitoxantrona, ou alguns tratamentos de câncer comumente usados.
- Medicamentos que afetam os rins, incluindo alguns antibióticos (usados para tratar infecções), diuréticos, alguns tipos de analgésicos (como o ibuprofeno e outros anti-inflamatórios semelhantes e medicamentos comprados sem receita médica) e medicamentos que contêm lítio .
- As vacinas administradas durante o tratamento com Tecfidera podem ser menos eficazes do que o normal. Tomar Tecfidera com certos tipos de vacinas (vacinas vivas) pode causar-lhe a infecção e, por conseguinte, deve ser evitado.
Tecfidera com comida, bebida e álcool
O consumo de bebidas com alto teor de álcool (mais de 30% de álcool por volume, por exemplo, licores) acima de uma pequena quantidade (mais de 50 ml) deve ser evitado dentro de uma "hora após tomar Tecfidera, porque o" álcool pode interagir com este medicamento. Isso pode causar inflamação do estômago (gastrite), principalmente em pessoas já com tendência à gastrite.
Avisos É importante saber que:
Gravidez e amamentação
Se está grávida, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Gravidez
Não use Tecfidera se estiver grávida, a menos que tenha discutido isso com seu médico
Hora da alimentação
Não se sabe se os ingredientes de Tecfidera passam para o leite materno. Tecfidera não deve ser utilizado durante a amamentação. O seu médico irá ajudá-la a decidir se deve interromper a amamentação ou o tratamento com Tecfidera. Isto pondera o benefício da amamentação para o seu bebé versus o benefício da terapêutica para ela.
Condução e utilização de máquinas
Não é conhecido o efeito de Tecfidera na capacidade de conduzir ou utilizar máquinas.O seu médico dir-lhe-á se a sua doença lhe permite conduzir e utilizar máquinas com segurança.
Dose, Método e Tempo de Administração Como usar Tecfidera: Posologia
Tome este medicamento sempre de acordo com as indicações do seu médico. Em caso de dúvida, consulte o seu médico ou farmacêutico.
Dose inicial
120 mg duas vezes ao dia.
Tome esta dose inicial durante os primeiros 7 dias e, em seguida, tome a dose regular
Dose regular
240 mg duas vezes ao dia.
Engula cada cápsula inteira com um pouco de água. Você não deve dividir, esmagar, dissolver, chupar ou mastigar a cápsula, pois isso pode aumentar alguns efeitos indesejados.
Tome Tecfidera com alimentos - pode ajudar a reduzir alguns dos efeitos secundários mais comuns (listados na secção 4).
Sobredosagem O que fazer se você tiver tomado muito Tecfidera
Se você tomar mais Tecfidera do que deveria
Se você tomou cápsulas demais, entre em contato com o seu médico imediatamente.
Se você esquecer de tomar Tecfidera
Se se esquecer ou se esquecer de uma dose, não tome uma dose a dobrar para compensar uma dose esquecida.
Você pode tomar a dose esquecida se você deixar pelo menos 4 horas entre as doses. Caso contrário, espere até a próxima dose programada.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
Efeitos colaterais Quais são os efeitos colaterais de Tecfidera
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
Efeitos colaterais graves
Níveis muito baixos de linfócitos - O número de linfócitos (um tipo de glóbulo branco) pode diminuir durante um longo período de tempo. A manutenção de níveis baixos de glóbulos brancos por um longo período de tempo pode aumentar o risco de infecções, incluindo o risco de uma infecção cerebral rara chamada leucoencefalopatia multifocal progressiva (PML). Os sintomas de PML podem ser semelhantes aos de uma recaída de EM. Os sintomas podem incluir o início ou agravamento da fraqueza em um lado do corpo (hemiparesia); má coordenação; mudanças na visão, pensamento ou memória; ou confusão ou alterações de personalidade que duram mais do que alguns dias.
- Se você tiver algum desses sintomas, chame seu médico imediatamente.
Reações alérgicas - não são comuns e podem afetar até 1 em 100 pessoas
Vermelhidão do rosto ou do corpo (rubor) é um efeito colateral muito comum (pode afetar mais de 1 em cada 10 pessoas). No entanto, se você sofre de vermelhidão e tem algum destes sinais:
- inchaço da face, lábios, boca ou língua
- respiração ofegante, dificuldade em respirar ou falta de ar
Pare de tomar Tecfidera e chame um médico imediatamente.
Efeitos colaterais muito comuns
Isso pode afetar mais de 1 em 10 pessoas:
- vermelhidão do rosto ou corpo, sensação de calor, calor, queimação ou coceira (rubor)
- fezes moles (diarreia)
- sensação de vômito iminente (náusea)
- dor de estômago ou cólicas estomacais
Tomar o medicamento com alimentos pode ajudar a reduzir os efeitos colaterais mencionados acima.
A presença de substâncias chamadas cetonas, que são produzidas naturalmente pelo corpo, é muito comumente revelada em testes de urina durante o tratamento com Tecfidera.
Converse com seu médico para obter informações sobre como controlar esses efeitos colaterais. Seu médico pode reduzir sua dose. Não reduza a dose a menos que o seu médico lhe diga para o fazer.
Efeitos colaterais comuns
Isso pode afetar até 1 em cada 10 pessoas:
- inflamação do revestimento do intestino (gastroenterite)
- Ele vomitou
- indigestão (dispepsia)
- inflamação do revestimento do estômago (gastrite)
- doença gastrointestinal
- sensação de queimadura
- afrontamento, sensação de calor
- pele com coceira
- irritação na pele
- manchas rosa ou vermelhas na pele (eritema)
Efeitos colaterais comuns, que podem aparecer em exames de sangue ou urina
- baixo nível de glóbulos brancos (linfocitopenia, leucopenia) no sangue. A redução no número de glóbulos brancos pode indicar que seu corpo é menos capaz de lutar contra uma "infecção. Se você tiver uma" infecção grave (como pneumonia), consulte seu médico imediatamente.
- proteína (albumina) na urina
- níveis aumentados de enzimas hepáticas (alanina aminotransferase, ALT e aspartato aminotransferase, AST) no sangue
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico, incluindo quaisquer efeitos secundários possíveis não mencionados neste folheto. Você também pode relatar os efeitos colaterais diretamente através do sistema nacional de notificação listado no Apêndice V. Ao relatar os efeitos colaterais, você pode ajudar a fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior após “VAL”. A data de validade refere-se ao último dia desse mês.
Não armazene acima de 30 ° C.
Conservar na embalagem original para proteger o medicamento da luz.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
O que Tecfidera contém
O ingrediente ativo é fumarato de dimetila. Tecfidera 120 mg: Cada cápsula contém 120 mg de fumarato de dimetila.
Tecfidera 240 mg: Cada cápsula contém 240 mg de fumarato de dimetilo.
Os outros ingredientes são celulose microcristalina, croscarmelose de sódio, talco, sílica coloidal anidra, estearato de magnésio, citrato de trietila, copolímero de ácido metacrílico-metacrilato de metila (1: 1), copolímero de ácido metacrílico-acrilato de etil (1: 1) 30%, simeticone , laurilsulfato de sódio, polissorbato 80, gelatina, dióxido de titânio (E171), Azul Brilhante FCF (E133), óxido de ferro amarelo (E172), goma laca, hidróxido de potássio e óxido de ferro preto (E172).
Descrição da aparência de Tecfidera e conteúdo da embalagem
As cápsulas gastrorresistentes de Tecfidera 120 mg são verdes e brancas com a impressão de 'BG-12 120 mg' e estão disponíveis em embalagens de 14 cápsulas.
As cápsulas gastrorresistentes de Tecfidera 240 mg são verdes e têm a impressão de 'BG-12 240 mg' e estão disponíveis em embalagens de 56 ou 168 cápsulas.
Nem todos os tamanhos de embalagem podem ser comercializados.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
TECFIDERA 120 - 240 MG CÁPSULAS GASTRORESISTANTES DURAS.
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Cápsula de Tecfidera 120 mg
Cada cápsula contém 120 mg de fumarato de dimetila.
Cápsula de Tecfidera 240 mg
Cada cápsula contém 240 mg de fumarato de dimetil
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Cápsula gastrorresistente dura
Cápsula de Tecfidera 120mg
Cápsula gastro-resistente verde e branca, impressa com “BG-12 120 mg”.
Cápsula de Tecfidera 240 mg
Cápsula verde, dura, gastro-resistente, impressa com "BG-12 240 mg"
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
Tecfidera está indicado para o tratamento de doentes adultos com esclerose múltipla recorrente-remitente (ver secção 5.1 para informações importantes sobre as populações para as quais a eficácia foi demonstrada).
04.2 Posologia e método de administração
O tratamento deve ser iniciado sob a supervisão de um médico com experiência no tratamento da doença.
Dosagem
A dose inicial é de 120 mg duas vezes ao dia. Após 7 dias, a dose é aumentada para a dose recomendada de 240 mg duas vezes ao dia.
A redução temporária da dose para 120 mg duas vezes ao dia pode reduzir o aparecimento de rubor e reações adversas gastrointestinais.Ao longo de 1 mês, a dose recomendada de 240 mg duas vezes ao dia deve ser retomada.
Tecfidera deve ser tomado com alimentos (ver secção 5.2). Tomar Tecfidera com alimentos pode melhorar a tolerabilidade em doentes que podem ter tendência a vermelhidão ou reações adversas gastrointestinais (ver secções 4.4, 4.5 e 4.8).
Cidadãos idosos
Os estudos clínicos de Tecfidera incluíram um número limitado de doentes com idade igual ou superior a 55 anos e não incluíram um número suficiente de doentes com 65 ou mais anos para determinar se respondem de forma diferente dos doentes mais jovens (ver secção 5.2). Com base no mecanismo de ação da substância ativa, não existe nenhuma razão teórica para a necessidade de ajustes de dose em idosos.
Insuficiência renal e hepática
Tecfidera não foi estudado em doentes com compromisso renal ou hepático. Com base em estudos de farmacologia clínica, não são necessários ajustes de dose (ver secção 5.2). Recomenda-se precaução no tratamento de doentes com compromisso renal ou compromisso hepático grave (ver secção 4.4).
População pediátrica
A segurança e eficácia de Tecfidera em crianças e adolescentes com idade entre 10 e 18 anos não foram ainda estabelecidas.Não existem dados disponíveis. Não existe utilização relevante de Tecfidera em crianças com menos de 10 anos para “Indicação de esclerose múltipla recorrente-remitente.
Método de administração
Para uso oral.
A cápsula ou o seu conteúdo não devem ser esmagados, divididos, dissolvidos, chupados ou mastigados, uma vez que o revestimento entérico dos microcomprimidos evita efeitos irritantes no intestino.
04.3 Contra-indicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na seção 6.1.
04.4 Advertências especiais e precauções adequadas de uso
Exames de sangue / análises laboratoriais
Foram observadas alterações laboratoriais na função renal e hepática em estudos clínicos em indivíduos tratados com Tecfidera (ver secção 4.8). As implicações clínicas dessas mudanças são desconhecidas. Avaliações da função renal (por exemplo, creatinina, valores de nitrogênio da ureia no sangue e urinálise) e função hepática (por exemplo, alanina aminotransferase, ALT e aspartato aminotransferase (AST) são recomendados antes do início da terapia, após 3 e 6 meses de terapia e a cada 6-12 meses a partir de então, conforme indicado clinicamente.
Os doentes tratados com Tecfidera podem desenvolver linfopenia grave e prolongada (ver secção 4.8). Tecfidera não foi estudado em doentes com contagens baixas de linfócitos pré-existentes e deve ter-se cuidado quando estes doentes são tratados. Antes de iniciar o tratamento com Tecfidera, deve ser feito um hemograma completo atualizado, incluindo linfócitos. Se a contagem de linfócitos for encontrada abaixo do intervalo normal, uma avaliação cuidadosa das possíveis causas deve ser realizada antes do início da terapia com Tecfidera.
Após o início da terapia, um hemograma completo, incluindo linfócitos, deve ser avaliado a cada 3 meses. Em pacientes com contagens de linfócitos
As contagens de linfócitos devem ser monitoradas até que se recuperem. Após a recuperação e na ausência de opções alternativas de tratamento, as decisões de reiniciar ou não Tecfidera após a descontinuação do tratamento devem ser baseadas na avaliação clínica.
Imagem de ressonância magnética (MRI)
Antes de iniciar o tratamento com Tecfidera, uma ressonância magnética de base (geralmente dentro de 3 meses) deve estar disponível para uso como referência. A necessidade de exames adicionais de imagem por ressonância magnética (MRI) deve ser avaliada de acordo com as recomendações nacionais e locais. A ressonância magnética (imagem MRI) pode ser considerada como parte do aumento do estado de alerta em pacientes considerados de risco aumentado para PML. Se houver suspeita clínica de PML, uma ressonância magnética deve ser realizada imediatamente para fins de diagnóstico.
Leucoencefalopatia multifocal progressiva (PML)
Com Tecfidera e outros produtos contendo fumarato, houve casos de PML no "cenário de linfopenia grave e prolongada. PML é uma infecção oportunista causada pelo vírus de John Cunningham (JCV), que pode ser fatal ou resultar em incapacidade grave... A leucoencefalopatia multifocal progressiva só pode ocorrer na presença de uma infecção por JCV. Ao testar JCV, deve-se levar em consideração que a influência da linfopenia na precisão do teste de anticorpos anti-JCV não foi estudada em pacientes tratados. tenha em mente que um teste negativo para a presença de anticorpos anti-JCV (na presença de contagens de linfócitos normais) não exclui a possibilidade de infecção pelo JCV no futuro.
Tratamento prévio com terapias imunossupressoras ou imunomoduladoras
Não foram realizados estudos para avaliar a eficácia e segurança de Tecfidera em doentes que mudaram de outras terapêuticas modificadoras da doença para Tecfidera.A contribuição de terapêuticas imunossupressoras anteriores no desenvolvimento de PML em doentes tratados com Tecfidera é desconhecida.Quando os pacientes mudam de outra terapia modificadora da doença para Tecfidera, a meia-vida e o modo de ação da outra terapia devem ser levados em consideração para evitar um efeito aditivo no sistema imunológico e, ao mesmo tempo, reduzir o risco de EM reativação.
Recomenda-se um hemograma completo antes de iniciar o tratamento com Tecfidera e em intervalos regulares durante o tratamento (ver Exames de sangue / análises laboratoriais sobre).
Geralmente, a terapia com Tecfidera pode ser iniciada imediatamente após a interrupção do interferon ou acetato de glatirâmero.
Insuficiência renal e hepática grave
Tecfidera não foi estudado em doentes com compromisso renal grave ou compromisso hepático grave e, portanto, deve-se ter precaução nestes doentes (ver secção 4.2).
Doença gastrointestinal ativa grave
Tecfidera não foi estudado em pacientes com doença gastrointestinal ativa grave e, portanto, deve-se ter cuidado nesses pacientes.
Vermelhidão (rubor)
Em estudos clínicos, 34% dos pacientes tratados com Tecfidera apresentaram vermelhidão. Na maioria dos pacientes com vermelhidão, esta foi de grau leve ou moderado.
Em estudos clínicos, 3 de um total de 2.560 doentes tratados com Tecfidera apresentaram sintomas graves de rubor, possivelmente atribuíveis a hipersensibilidade ou reações anafilactoides. Estes acontecimentos não foram potencialmente fatais, mas necessitaram de hospitalização.Os prescritores e os doentes devem estar cientes desta possibilidade em caso de reacções de rubor graves (ver secções 4.2, 4.5 e 4.8).
Infecções
Nos estudos de fase III controlados por placebo, a incidência de infecções (60% versus 58%) e infecções graves (2% versus 2%) foi semelhante em pacientes tratados com Tecfidera ou placebo, respectivamente. de infecções graves em doentes com contagens de linfócitos em estabilização (ver secção 4.8). A contagem média de linfócitos permaneceu dentro dos limites normais. Contagem de linfócitos
Se a terapia for continuada na presença de linfopenia grave e prolongada, o risco de infecções oportunistas, incluindo linfopenia, não pode ser excluído.eukoencefalopatia multifocal progressiva (PML) (consulte a subseção sobre PML acima para obter mais detalhes).
Se um paciente desenvolver uma infecção grave, a interrupção de Tecfidera deve ser considerada e os benefícios e riscos reavaliados antes de reiniciar a terapia. Os pacientes que tomam Tecfidera devem ser aconselhados a relatar os sintomas de infecções ao médico. ) foram resolvidos.
04.5 Interações com outros medicamentos e outras formas de interação
Tecfidera não foi estudado em combinação com terapias antineoplásicas ou imunossupressoras e, portanto, deve-se ter cuidado durante a administração concomitante. Em ensaios clínicos de esclerose múltipla, o tratamento concomitante de recidivas com um ciclo curto de corticosteróides intravenosos não foi associado a um aumento clinicamente relevante da infecção.
A vacinação durante o tratamento com Tecfidera não foi avaliada. Não se sabe se o tratamento com Tecfidera reduzirá a eficácia de algumas vacinas. As vacinas vivas podem acarretar um risco aumentado de infecção clínica e não devem ser administradas a pacientes tratados com Tecfidera, a menos que, em casos excepcionais, este risco potencial seja considerado menos importante do que o risco de não vacinação para o indivíduo.
Durante o tratamento com Tecfidera, o uso concomitante de outros derivados do ácido fumárico (tópico ou sistémico) deve ser evitado.
Em humanos, o fumarato de dimetila é extensivamente metabolizado pelas esterases antes de atingir a circulação sistêmica e ocorre metabolismo posterior através do ciclo do ácido tricarboxílico, sem qualquer envolvimento do sistema do citocromo P450 (CYP). Nenhum risco potencial de interações medicamentosas foi identificado a partir dos estudos em vitro inibição e indução de CYP, a partir de um estudo de p-glicoproteínas ou de estudos de ligação a proteínas de fumarato de dimetila e fumarato de monometila (um metabólito primário de fumarato de dimetila).
Os medicamentos comumente usados em pacientes com esclerose múltipla, como interferon beta-1a e acetato de glatirâmero administrados por via intramuscular, foram testados clinicamente para potenciais interações com fumarato de dimetila e não alteraram o perfil farmacocinético do fumarato de dimetila.
Em um estudo realizado em voluntários saudáveis, a administração de 325 mg (ou equivalente) de ácido acetilsalicílico com revestimento não entérico, 30 minutos antes de Tecfidera, ao longo de 4 dias de administração, não alterou o perfil farmacocinético de Tecfidera e reduziu o “Início e gravidade do rubor. No entanto, o uso a longo prazo de ácido acetilsalicílico não é recomendado para o tratamento da vermelhidão. Os riscos potenciais associados à terapia com ácido acetilsalicílico devem ser considerados antes da administração concomitante com Tecfidera (ver secções 4.2, 4.4 e 4.8).
A terapia concomitante com medicamentos nefrotóxicos (tais como aminoglicosídeos, diuréticos, AINEs ou lítio) pode aumentar potenciais reações adversas renais (por exemplo, proteinúria) em pacientes tratados com Tecfidera (ver secção 4.8).
O consumo de quantidades moderadas de álcool não alterou a exposição a Tecfidera e não foi associado a um aumento nas reações adversas. O consumo de grandes quantidades de bebidas com alto teor alcoólico (mais de 30% de álcool por volume) pode resultar em um aumento nas taxas de dissolução de Tecfidera e pode, portanto, aumentar a frequência das reações adversas gastrointestinais.
Estudos em vitro A indução do CYP não demonstrou interação entre Tecfidera e contraceptivos orais. Nenhum estudo foi realizado na Vivo sobre interação com contraceptivos orais Embora não seja esperada nenhuma interação, devem ser consideradas medidas contraceptivas não hormonais com Tecfidera (ver secção 4.6).
População pediátrica
Os estudos de interação foram realizados apenas em adultos.
04.6 Gravidez e lactação
Gravidez
Não existem dados ou existem quantidades limitadas de dados sobre a utilização de fumarato de dimetilo em mulheres grávidas. Os estudos em animais revelaram toxicidade reprodutiva (ver secção 5.3). Tecfidera não é recomendado durante a gravidez e em mulheres com potencial para engravidar. Sem utilizar contraceptivos adequados ( ver secção 4.5) Tecfidera só deve ser utilizado durante a gravidez se for claramente necessário e se o potencial benefício justificar o potencial risco para o feto.
Hora da alimentação
Não se sabe se o fumarato de dimetila ou seus metabólitos são excretados no leite humano. Um risco para os recém-nascidos / bebês não pode ser excluído. Deve ser tomada a decisão de interromper a amamentação ou interromper a terapia com Tecfidera.O benefício da amamentação para a criança e o benefício da terapia para a mulher devem ser considerados.
Fertilidade
Não existem dados sobre os efeitos de Tecfidera na fertilidade humana. Os dados fornecidos pelos estudos pré-clínicos não sugerem que o fumarato de dimetilo esteja associado a um risco aumentado de diminuição da fertilidade (ver secção 5.3).
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
Não foram realizados estudos sobre a capacidade de conduzir e utilizar máquinas.
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
As reações adversas mais comuns (incidência ≥10%) em doentes tratados com Tecfidera foram rubor (rubor) e eventos gastrointestinais (isto é, diarreia, náusea, dor abdominal, dor abdominal superior). Rubor e eventos gastrointestinais tendem a surgir no início da terapia (especialmente durante o primeiro mês) e em pacientes com tendência a vermelhidão e eventos gastrointestinais, esses eventos podem continuar a ocorrer intermitentemente durante o tratamento com Tecfidera. Reações adversas relatadas mais comumente levando à descontinuação da terapia ( incidência> 1%) em pacientes tratados com Tecfidera apresentaram rubor (3%) e eventos gastrointestinais (4%).
Em ensaios clínicos controlados por placebo e não controlados, um total de 2.468 doentes receberam Tecfidera e acompanhados até 4 anos com uma exposição global equivalente a 3.588 pessoas-ano. Aproximadamente 1.056 doentes receberam mais de 2 anos de terapêutica. Com Tecfidera. Experiência em os ensaios clínicos não controlados são consistentes com a experiência em ensaios clínicos controlados com placebo.
Tabela de reações adversas
A tabela abaixo mostra as reações adversas notificadas com maior frequência em doentes tratados com Tecfidera do que em doentes tratados com placebo. Estes dados são derivados de 2 ensaios clínicos principais de Fase 3, duplo-cegos, controlados por placebo com um total de 1.529 doentes tratados com Tecfidera até 24 meses, com uma exposição global de 2.371 pessoas-ano (ver secção 5.1. Frequências descritas na tabela abaixo, baseiam-se em 769 pacientes tratados com Tecfidera 240 mg duas vezes ao dia e 771 pacientes tratados com placebo.
As reações adversas são apresentadas de acordo com a terminologia recomendada MedDRA na Classe de Sistemas de Órgãos MedDRA. A incidência das reações adversas listadas abaixo é expressa de acordo com a seguinte convenção:
- Muito comum (≥1 / 10)
- Comum (≥1 / 100,
- Incomum (≥1 / 1.000,
- Raro (≥1 / 10.000,
- Muito raro (
- Desconhecido (a frequência não pode ser estimada a partir dos dados disponíveis)
Descrição das reações adversas selecionadas
Vermelhidão (rubor)
Em ensaios clínicos controlados com placebo, a incidência de vermelhidão (rubor) (34% vs 4%) e afrontamentos (7% vs 2%) aumentaram nos doentes tratados com Tecfidera em comparação com os tratados com placebo, respetivamente. O rubor é tipicamente descrito como vermelhidão ou ondas de calor, mas pode incluir outros eventos (por exemplo, calor, vermelhidão, coceira e sensação de queimação). Os eventos de rubor tendem a ocorrer no início da terapia (especialmente durante o primeiro mês) e, em pacientes afetados, esses eventos podem continuar a ocorrer de forma intermitente durante o tratamento com Tecfidera. Em pacientes com rubor, a maioria apresentou eventos de rubor leves ou moderados. No geral, 3% dos doentes tratados com Tecfidera descontinuaram o tratamento devido ao rubor. A incidência de rubor grave, que pode ser caracterizado por eritema generalizado, erupção cutânea e / ou prurido, foi observada em menos de 1% dos doentes tratados com Tecfidera (ver secções 4.2, 4.4 e 4.5).
Gastrointestinal
A incidência de eventos gastrointestinais (por exemplo, diarreia [14% vs 10%], náusea [12% vs 9%], dor abdominal superior [10% vs 6%], dor abdominal [9% vs 4%], vômitos [8 % versus 5%] e dispepsia [5% versus 3%]) aumentaram em pacientes tratados com Tecfidera em comparação com aqueles tratados com placebo, respectivamente. Os eventos gastrointestinais tendem a ocorrer no início da terapia (especialmente durante o primeiro mês) e, nos pacientes afetados doentes, estes acontecimentos podem continuar a ocorrer de forma intermitente ao longo do tratamento com Tecfidera. Na maioria dos pacientes que apresentaram eventos gastrointestinais, estes foram de gravidade leve ou moderada. 4% dos pacientes tratados com Tecfidera interromperam a terapia devido a eventos gastrointestinais. A incidência de acontecimentos gastrointestinais graves, incluindo gastroenterite e gastrite, foi observada em 1% dos doentes tratados com Tecfidera (ver secção 4.2).
Transaminases hepáticas
Em estudos controlados com placebo, foram observados aumentos nas transaminases hepáticas. Na maioria dos pacientes nos quais essas elevações ocorreram, as transaminases hepáticas foram alanina aminotransferase e aspartato aminotransferase (AST) ≥3 vezes LSN, foram observadas em 5% e 2% dos pacientes tratados com placebo, respectivamente. E em 6% e 2% de doentes tratados com Tecfidera. Não foram observados aumentos das transaminases ≥3 vezes LSN com aumentos concomitantes na bilirrubina total> 2 vezes LSN. Descontinuação da terapêutica devido a transaminases hepáticas elevadas.
Renal
Em estudos controlados com placebo, a incidência de proteinúria foi maior em pacientes tratados com Tecfidera (9%) em comparação com placebo (7%). A incidência geral de eventos adversos renais e urinários foi semelhante para pacientes tratados com Tecfidera e com placebo.Nenhum caso de insuficiência renal grave foi relatado. A análise de urina mostra que a porcentagem de pacientes com valores de proteína de 1+ ou superior é semelhante para pacientes tratados com Tecfidera (43%) e pacientes tratados com placebo (40%). Normalmente, as observações laboratoriais de proteinúria não foram progressivas. Em comparação com doentes tratados com placebo, foi observado um aumento na taxa de filtração glomerular estimada (eTFG) em doentes tratados com Tecfidera, incluindo aqueles que experimentaram 2 episódios consecutivos de proteinúria (≥1 +).
Hematológico
Em ensaios clínicos controlados com placebo, os valores dos linfócitos eram normais na maioria dos doentes (> 98%) antes do início da terapêutica. Assim que o tratamento com Tecfidera foi iniciado, as contagens médias de linfócitos diminuíram ao longo do primeiro ano e, subsequentemente, estabilizaram. Em média, as contagens de linfócitos diminuíram aproximadamente 30% em relação ao valor basal. As contagens de linfócitos média e mediana permaneceram dentro dos limites normais. A contagem de linfócitos eosinofílicos foi observada durante os primeiros 2 meses de terapia.
Anormalidades laboratoriais
Em ensaios clínicos controlados com placebo, as medições de cetonas na urina (1+ ou superior) foram superiores em doentes tratados com Tecfidera (45%) em comparação com o placebo (10%). Nenhuma consequência inesperada foi observada em estudos clínicos.
Os níveis de 1,25-diidroxivitamina D diminuíram em pacientes tratados com Tecfidera em comparação com aqueles tratados com placebo (diminuição da porcentagem média desde o início até 2 anos em 25% em comparação com 15%, respectivamente) e os níveis de hormônio da paratireóide (PTH) aumentaram em pacientes tratados com Tecfidera em comparação com aqueles tratados com placebo (aumento na porcentagem média desde o início para 2 anos de 29% em comparação com 15%, respectivamente). Os valores médios para ambos os parâmetros permaneceram dentro da faixa normal.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas que ocorram após a autorização do medicamento é importante, uma vez que permite a monitorização contínua da relação benefício / risco do medicamento.Os profissionais de saúde são convidados a notificar quaisquer suspeitas de reações adversas através da Agência Italiana de Medicamentos. . Site: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdose
Nenhum caso de sobredosagem foi relatado.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: outros medicamentos para o sistema nervoso.
Código ATC: N07XX09.
Mecanismo de ação
O mecanismo pelo qual o fumarato de dimetila exerce efeitos terapêuticos na esclerose múltipla não é totalmente compreendido. Estudos pré-clínicos indicam que as respostas farmacodinâmicas do fumarato de dimetila são mediadas principalmente pela ativação da via de transcrição do fator nuclear Nrf2 (fator nuclear eritróide 2 2 relacionado). O fumarato de dimetila mostrou causar regulação positiva em pacientes (suprarregulação) de genes antioxidantes dependentes de Nrf2 (por exemplo, NAD (P) H desidrogenase, quinona 1; [NQO1]).
Efeitos farmacodinâmicos
Efeitos no sistema imunológico
Em estudos pré-clínicos e clínicos, Tecfidera demonstrou propriedades anti-inflamatórias e imunomoduladoras. O fumarato de dimetila e o fumarato de monometila, o principal metabólito do fumarato de dimetila, reduziram significativamente a ativação das células imunológicas e a liberação subsequente de citocinas pró-inflamatórias em resposta a estímulos inflamatórios em modelos pré-clínicos. Em estudos clínicos em pacientes com psoríase, o fumarato de dimetila afetou os fenótipos dos linfócitos por regulação baixa (desregulação) de perfis de citocinas pró-inflamatórias (TH1, TH17) e favoreceu a produção de citocinas anti-inflamatórias (TH2). O fumarato de dimetila demonstrou atividade terapêutica em vários modelos de lesões inflamatórias e neuroinflamatórias. Em estudos de Fase 3, durante o tratamento com Tecfidera, a contagem média de linfócitos diminuiu em uma média de cerca de 30% da linha de base durante o primeiro ano, com uma fase de estabilização subsequente.
Efeito no sistema cardiovascular
Num estudo de intervalo QT corrigido (QTc), doses únicas de Tecfidera 240 mg ou 360 mg comparadas com placebo não tiveram efeito no intervalo QTc.
Eficácia clínica e segurança
Dois estudos randomizados, duplo-cegos, controlados por placebo, de 2 anos foram conduzidos [Estudo 1 (DEFINE) com 1.234 indivíduos e Estudo 2 (CONFIRM) com 1.417 indivíduos] em indivíduos com esclerose múltipla recorrente-remitente (MS -RR). Nenhum indivíduo com formas progressivas de esclerose múltipla foi incluído nestes estudos. A eficácia (ver tabela abaixo) e a segurança foram demonstradas em indivíduos com escores da Escala Expandida de Status de Incapacidade (EDSS) variando de 0 a 5, inclusive, que tiveram pelo menos 1 recaída durante "ano antes da randomização ou, dentro de 6 semanas de randomização, eles teve imagem de ressonância magnética do cérebro (MRI) mostrando pelo menos uma lesão de realce de gadolínio (Gd +). O estudo 2 incluiu um braço comparador simples-cego (cego pelo avaliador, ou seja, o médico / investigador do estudo avaliando a resposta ao tratamento no estudo estava em uma condição cega) de tratamento com acetato de glatirâmero (GA).
No Estudo 1, os pacientes apresentavam as seguintes características basais medianas: idade 39 anos, duração da doença 7,0 anos, pontuação EDSS 2,0. Além disso, 16% dos pacientes tiveram uma pontuação de EDSS> 3,5, 28% tiveram ≥2 recidivas no ano anterior e 42% haviam recebido outros tratamentos aprovados para esclerose múltipla. Na coorte de ressonância magnética, 36% dos pacientes incluídos no estudo tinha lesões que aumentam o gadolínio (Gd +) na linha de base (número médio de lesões Gd + 1,4).
No Estudo 2, os pacientes apresentavam as seguintes características basais: idade 37 anos, duração da doença 6,0 anos, pontuação EDSS 2,5. Além disso, 17% dos pacientes tiveram uma pontuação de EDSS> 3,5, 32% tiveram ≥2 recidivas no ano anterior e 30% haviam recebido outros tratamentos aprovados para esclerose múltipla. Na coorte de ressonância magnética, 45% dos pacientes incluídos no estudo teve lesões que aumentam o gadolínio (Gd +) na linha de base (número médio de lesões Gd + 2,4).
Em comparação com o placebo, os indivíduos tratados com Tecfidera tiveram uma redução clinicamente relevante e estatisticamente significativa em: a proporção de indivíduos com recidiva aos 2 anos, parâmetro de avaliação primário do Estudo 1; a taxa de recaída anualizada de 2 anos, desfecho primário do Estudo 2.
A taxa de recaída anual para acetato de glatirâmero e placebo foi de 0,286 e 0,401 no Estudo 2, respectivamente, correspondendo a uma redução de 29% (p = 0,013), o que é consistente com a informação de prescrição aprovada.
a Todas as análises de desfechos clínicos foram por intenção de tratar (ITT);
b A análise de ressonância magnética usou a coorte de ressonância magnética
* Valor P
Eficácia em pacientes com alta atividade da doença:
Um efeito consistente do tratamento na recidiva foi observado em um subgrupo de pacientes com alta atividade da doença, enquanto o efeito no tempo para a progressão sustentada da incapacidade em 3 meses não foi claramente estabelecido. Devido ao desenho do estudo, a "alta atividade da doença foi definida do seguinte modo:
- Pacientes com 2 ou mais recidivas em um ano e com uma ou mais lesões potencializadoras de Gadolínio (Gd) na ressonância magnética (MRI) do cérebro (n = 42 no estudo DEFINE; n = 51 no estudo CONFIRM) ou ,
- Pacientes que não responderam a um curso completo e adequado (pelo menos um ano de tratamento) de interferon beta, tendo tido pelo menos 1 recaída no ano anterior na terapia e pelo menos 9 lesões T2 hiperintensas na ressonância magnética (MRI ) do crânio ou pelo menos uma lesão que realça o gadolínio (Gd), ou pacientes com taxa de recidiva inalterada ou maior no ano anterior em comparação com os 2 anos anteriores (n = 177 no estudo DEFINE; n = 141 no CONFIRM estude).
População pediátrica
A Agência Europeia de Medicamentos diferiu a obrigação de apresentação dos resultados dos estudos com Tecfidera em um ou mais subgrupos da população pediátrica na esclerose múltipla (ver secção 4.2 para informação sobre utilização pediátrica).
05.2 "Propriedades farmacocinéticas
Administrado por via oral, Tecfidera (fumarato de dimetila) sofre rápida hidrólise pré-sistêmica mediada por esterase e é convertido em fumarato de monometila, seu principal metabólito, que também é ativo. Fumarato de dimetila não é quantificável no plasma após a administração oral de Tecfidera. Portanto, todas as análises farmacocinéticas relacionadas ao fumarato de dimetila foram realizadas com concentrações plasmáticas de fumarato de monometila Os dados farmacocinéticos foram obtidos em indivíduos com esclerose múltipla e em voluntários saudáveis.
Absorção
O Tmax do fumarato de monometila é entre 2 e 2,5 horas. Uma vez que Tecfidera cápsulas gastrorresistentes contém microcápsulas, que são protegidas por um revestimento entérico, a absorção não começa até que saiam do estômago (normalmente menos de 1 hora). Após a administração com alimentos de 240 mg duas vezes por dia, a mediana o pico (Cmax) foi de 1,72 mg / le a exposição geral (AUC, área sob a curva) foi de 8,02 h.mg / l em indivíduos com esclerose múltipla. No geral, o C
max e AUC aumentaram aproximadamente proporcionalmente à dose ao longo do intervalo de dose estudado (120 mg a 360 mg). Em indivíduos com esclerose múltipla, duas doses de 240 mg foram administradas com 4 horas de intervalo durante um período de 4 horas. regime de administração de dosagem três vezes a dia. Isso resultou em um acúmulo mínimo de exposição, resultando em um aumento de 12% na Cmax mediana em comparação com a dosagem de duas vezes ao dia (1,72 mg / L duas vezes ao dia vs 1,93 mg / L três vezes ao dia), sem implicações de segurança.
Os alimentos não têm efeito clinicamente significativo na exposição ao fumarato de dimetilo.No entanto, Tecfidera deve ser tomado com alimentos devido à melhoria da tolerabilidade à vermelhidão ou aos acontecimentos adversos gastrointestinais (ver secção 4.2).
Distribuição
O volume aparente de distribuição após a administração oral de Tecfidera 240 mg varia entre 60 L e 90 L. A ligação do fumarato de monometilo às proteínas plasmáticas humanas é geralmente entre 27% e 40%.
Biotransformação
Em humanos, o fumarato de dimetila é extensivamente metabolizado com menos de 0,1% da dose excretada na urina como fumarato de dimetila não modificado. O fumarato de dimetila é inicialmente metabolizado por esterases, que são onipresentes no trato gastrointestinal, sangue e tecidos, antes de atingir a circulação sistêmica . O metabolismo posterior ocorre por meio do ciclo do ácido tricarboxílico, sem envolvimento do sistema do citocromo P450 (CYP). Um estudo de dose única de 240 mg de fumarato de 14C-dimetila identificou a glicose como o metabólito predominante no plasma humano. Outros metabólitos circulantes incluíram o ácido fumárico, ácido cítrico e fumarato de monometila.O metabolismo do ácido fumárico a jusante da referida via metabólica se dá por meio do ciclo do ácido tricarboxílico, com a exalação de dióxido de carbono (CO2) que atua como principal via de eliminação.
Eliminação
A exalação de CO2 é a principal via de eliminação do fumarato de dimetila e responde por 60% da dose.A eliminação renal e fecal são vias secundárias de eliminação, respondendo por 15,5% e 0,9% da dose, respectivamente.
A meia-vida terminal do fumarato de monometila é curta (aproximadamente 1 hora) e nenhum fumarato de monometila circulante está presente em 24 horas na maioria dos indivíduos. O acúmulo do fármaco original ou fumarato de monometila não ocorre com doses múltiplas de fumarato de dimetila no regime terapêutico.
Linearidade
A exposição ao fumarato de dimetilo aumenta de forma aproximadamente proporcional à dose com doses únicas e múltiplas ao longo do intervalo de doses estudado de 120 mg a 360 mg.
Farmacocinética em grupos de pacientes especiais
Com base nos resultados da análise de variância (ANOVA), o peso corporal é a principal covariável de exposição (de acordo com Cmax e AUC) em indivíduos com esclerose múltipla recorrente-remitente (RRMS), mas não afetou as medições. Segurança e eficácia avaliadas em ensaios clínicos.
O sexo e a idade não tiveram um impacto clinicamente significativo na farmacocinética do fumarato de dimetilo. A farmacocinética em doentes com 65 ou mais anos de idade não foi estudada.
População pediátrica
A farmacocinética em pacientes menores de 18 anos não foi estudada.
Insuficiência renal
Uma vez que a via renal é uma via secundária de eliminação para o fumarato de dimetila, representando menos de 16% da dose administrada, não foi realizada avaliação da farmacocinética em indivíduos com insuficiência renal.
Insuficiência hepática
Uma vez que o fumarato de dimetilo e o fumarato de monometilo são metabolizados por esterases, sem envolvimento do sistema CYP450, não foi realizada avaliação da farmacocinética em indivíduos com compromisso hepático.
05.3 Dados de segurança pré-clínica
As reações adversas descritas nas secções de Toxicologia e Toxicidade Reprodutiva abaixo não foram observadas em estudos clínicos, mas foram observadas em animais com níveis de exposição semelhantes aos níveis de exposição clínica.
Mutagênese
O fumarato de dimetila e o fumarato de mono-metila foram negativos em uma bateria de testes em vitro (Teste de Ames, teste de aberrações cromossômicas em células de mamíferos). Fumarato de dimetila foi negativo no teste de micronúcleo de rato na Vivo.
Carcinogênese
Os estudos de carcinogenicidade do fumarato de dimetila foram conduzidos por até 2 anos em camundongos e ratos. O fumarato de dimetila foi administrado por via oral em doses de 25, 75, 200 e 400 mg / kg / dia para camundongos e em doses de 25, 50, 100 e 150 mg / kg / dia para ratos. Em ratinhos, a incidência de carcinoma tubular renal aumentou com a dose de 75 mg / kg / dia, uma exposição equivalente (AUC) com a dose humana recomendada. Em ratos, a incidência de carcinoma tubular renal aumentou com uma dose de 100 mg / kg / dia, uma exposição aproximadamente 3 vezes a dose humana recomendada. A relevância dessas descobertas para o risco humano é desconhecida.
A incidência de papiloma e carcinoma de células escamosas na parte não glandular do estômago (antes do estômago) aumentou com uma exposição equivalente à dose humana recomendada em camundongos e com uma exposição abaixo da dose humana recomendada em ratos (com base em "AUC ) Não há contrapartida humana para o estômago de roedores.
Toxicologia
Os estudos pré-clínicos foram realizados em roedores, coelhos e macacos com uma suspensão de fumarato de dimetilo (fumarato de dimetilo em hidroxipropilmetilcelulose a 0,8%) administrado por sonda oral. O estudo crônico em cães foi conduzido com administração oral da cápsula de fumarato de dimetila.
Alterações renais foram observadas após administração oral repetida de fumarato de dimetila em camundongos, ratos, cães e macacos. Regeneração do epitélio tubular renal, indicativa de lesão, foi observada em todas as espécies.Hiperplasia tubular renal foi observada em ratos que receberam tratamento vitalício (estudo de 2 anos). Atrofia cortical foi observada em cães e macacos, e necrose de célula única e fibrose intersticial foram observadas em macacos que receberam doses orais diárias de fumarato de dimetila por 12 meses, em 6 vezes a dose recomendada com base na AUC. Sabe a relevância desses achados para humanos risco.
A degeneração do epitélio seminífero foi observada nos testículos de ratos e cães Os resultados foram observados com aproximadamente a dose recomendada em ratos e 6 vezes a dose recomendada em cães (com base na AUC). A relevância dessas descobertas para o risco humano é desconhecida.
Os achados no estômago anterior de camundongos e ratos foram hiperplasia epitelial escamosa associada a hiperqueratose; inflamação; e papiloma e carcinoma de células escamosas em estudos com duração de 3 meses ou mais. Não há contrapartida humana para o estômago anterior de camundongos e ratos.
Toxidade reprodutiva
A administração oral de fumarato de dimetila a ratos machos em 75, 250 e 375 mg / kg / dia antes e durante o acasalamento não teve efeito na fertilidade masculina até a dose mais alta testada (pelo menos 2x a dose AUC recomendada). A administração oral de fumarato de dimetila a ratas nas doses de 25, 100 e 250 mg / kg / dia antes e durante o acasalamento, e continuando até o dia 7 de gestação, resultou em uma redução no número de ciclos de estro por 14 dias e aumentou o número de animais em diestro prolongado na dose mais alta testada (11 vezes a dose recomendada com base na AUC). No entanto, essas mudanças não tiveram efeito sobre a fertilidade ou o número de fetos viáveis produzidos.
O fumarato de dimetila demonstrou atravessar a membrana placentária e entrar no sangue fetal de ratos e coelhos, com taxas de concentração plasmática fetal / materna variando de 0,48 a 0,64 e 0,1, respectivamente. Não foram observadas malformações em ratos ou coelhos com qualquer dose de fumarato de dimetila. A administração de fumarato de dimetila em doses orais de 25, 100 e 250 mg / kg / dia para ratas grávidas durante o período de organogênese produziu efeitos adversos maternos em 4 vezes a dose recomendada com base na AUC, baixo peso fetal e "ossificação retardada" (metatarsal) e falanges dos membros posteriores) com 11 vezes a dose recomendada com base na AUC. Peso fetal inferior e ossificação retardada foram considerados secundários à toxicidade materna (redução do peso corporal e consumo de alimentos).
A administração oral de fumarato de dimetila em 25, 75 e 150 mg / kg / dia para coelhas grávidas durante a organogênese não teve efeito sobre o desenvolvimento embriofetal e resultou na redução do peso materno para 7 vezes a dose recomendada e aumento do aborto. Para 16 vezes a dose recomendada, com base na AUC.
A administração oral de fumarato de dimetila a 25, 100 e 250 mg / kg / dia a ratos durante a gravidez e lactação resultou em pesos corporais reduzidos em ninhadas F1 e atrasos na maturação sexual em machos F1 com 11 vezes a dose recomendada com base em "AUC. Não houve efeito na fertilidade nas ninhadas F1. O peso corporal inferior das ninhadas foi considerado secundário à toxicidade materna.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Micro-comprimidos com revestimento entérico
Celulose microcristalina
Croscarmelose de sódio
Talco
Sílica coloidal anidra
Estearato de magnesio
Citrato de trietila
Ácido metacrílico - copolímero de metacrilato de metila (1: 1)
Dispersão de copolímero de ácido metacrílico - acrilato de etila (1: 1) 30%
Simeticone
Lauril sulfato de sódio
Polissorbato 80
Concha da cápsula
Geléia
Dióxido de titânio (E171)
FCF Azul Brilhante (E133)
Óxido de ferro amarelo (E172)
Impressão da cápsula (tinta preta)
Goma laca
Hidróxido de potássio
Óxido de ferro preto (E172)
06.2 Incompatibilidade
Não é relevante.
06.3 Período de validade
120 mg cápsulas gastrorresistentes: 4 anos.
Cápsulas gastrorresistentes de 240 mg: 3 anos.
06.4 Precauções especiais de armazenamento
Não armazene acima de 30 ° C.
Manter os blisters na embalagem exterior para proteger o medicamento da luz.
06.5 Natureza da embalagem primária e conteúdo da embalagem
Cápsulas de 120 mg: 14 cápsulas em embalagens de blister de PVC / PE / PVDC-PVC.
Cápsulas de 240 mg: 56 ou 168 cápsulas em embalagens blister de alumínio de PVC / PE / PVDC-PVC.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio
Sem instruções especiais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Biogen Idec Ltd
Casa da Inovação
70 Norden Road
Virgindade
Berkshire
SL6 4AY
Reino Unido
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
A.I.C. n. 043217013 / E
A.I.C. n. 043217025 / E
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 30 de janeiro de 2014
10.0 DATA DE REVISÃO DO TEXTO
12/2015