Ingredientes ativos: Capecitabina
Comprimidos revestidos por película de Xeloda 150 mg
As bulas de Xeloda estão disponíveis para os tamanhos de embalagem:- Comprimidos revestidos por película Xeloda 150 mg
- Comprimidos revestidos por película de Xeloda 500 mg
Por que o Xeloda é usado? Para que serve?
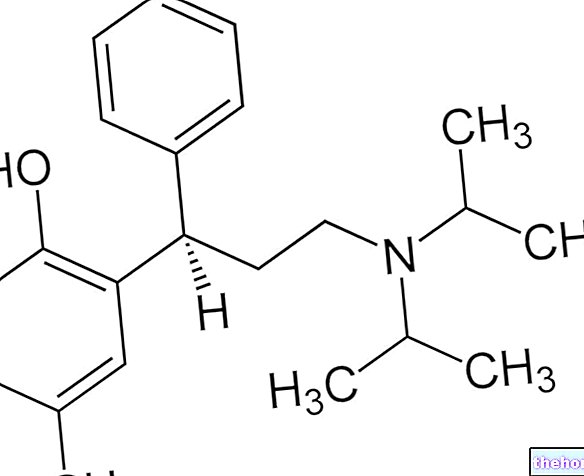
Xeloda pertence a um grupo de medicamentos denominados “medicamentos citostáticos”, que impedem o crescimento das células cancerígenas. Xeloda contém 150 mg de capecitabina, que por si só não é um medicamento citostático. Apenas uma vez absorvido pelo corpo é transformado em um medicamento anticâncer ativo (em maior extensão nos tecidos tumorais do que nos tecidos normais).
O Xeloda é prescrito por médicos para tratar cancros do cólon, reto, estômago ou mama. Além disso, o Xeloda é prescrito para prevenir o aparecimento de um novo cancro do cólon após a remoção cirúrgica completa do tumor.
Xeloda pode ser usado sozinho ou em combinação com outros medicamentos.
Contra-indicações Quando Xeloda não deve ser usado
Não tome Xeloda:
- se tem alergia à capecitabina ou a qualquer outro componente deste medicamento (listados na secção 6). Você deve informar o seu médico se souber que é alérgico ou tiver uma reação exagerada a este medicamento,
- se já teve uma reação grave anterior à terapia com fluoropirimidina (um grupo de medicamentos anticâncer como o fluorouracil),
- se você está grávida ou amamentando,
- se tiver níveis excessivamente baixos de glóbulos brancos e plaquetas no sangue (leucopenia, neutropenia ou trombocitopenia),
- se você tem problemas graves de fígado ou rins,
- se tiver uma deficiência conhecida da enzima dihidropirimidina desidrogenase (DPD) envolvida no metabolismo do uracilo e da timina ou
- se está a ser tratado ou foi tratado nas últimas 4 semanas com brivudina, sorivudina ou substâncias de classes semelhantes como parte da terapêutica para herpes zoster (catapora ou incêndio de Santo António).
Precauções de uso O que você precisa saber antes de tomar Xeloda
Fale com o seu médico ou farmacêutico antes de tomar Xeloda:
- se você tem doença renal ou hepática,
- se teve ou tem problemas cardíacos (por exemplo, frequência cardíaca irregular ou dores irradiando do peito para a mandíbula e vice-versa causadas por esforço físico e devido a problemas com o fluxo sanguíneo para o coração),
- se você tem doença cerebral (por exemplo, um tumor que se espalhou para o cérebro) ou lesão nervosa (neuropatia),
- se você tiver desequilíbrios nos níveis de cálcio (detectáveis em exames de sangue),
- se voce tem diabetes,
- se você não consegue manter comida ou água em seu corpo devido a náuseas e vômitos graves,
- se você sofre de diarréia,
- se você está ou pode ficar desidratado,
- se você tiver desequilíbrios de íons no sangue (desequilíbrios eletrolíticos, que podem ser encontrados em exames de sangue),
- se sofreu de problemas oculares, pois pode necessitar de monitorização ocular adicional.
- se tiver uma reação cutânea grave.
Deficiência de dihidropirimidina desidrogenase (DPD): a deficiência de DPD é uma doença rara presente ao nascimento que geralmente não está associada a problemas de saúde, a menos que certos medicamentos sejam tomados. Se tem uma deficiência de DPD desconhecida e está a tomar Xeloda, os efeitos secundários listados na secção 4 “Efeitos secundários possíveis” podem ocorrer de forma grave. Informe o seu médico se algum dos efeitos secundários o preocupa ou se notar quaisquer efeitos secundários não mencionados neste folheto (ver secção 4 “Efeitos secundários possíveis”).
Crianças e adolescentes
Xeloda não é indicado para o tratamento de crianças e adolescentes. Não dê Xeloda a crianças e adolescentes.
Interações Quais medicamentos ou alimentos podem alterar o efeito do Xeloda
Outros medicamentos e Xeloda
Antes de iniciar o tratamento, informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente ou se vier a tomar outros medicamentos. Isto é de fundamental importância, uma vez que a ingestão concomitante de vários medicamentos pode potencializar ou reduzir o seu efeito. Atenção especial deve ser dada no caso de ingestão concomitante de:
- medicamentos para a gota (alopurinol),
- medicamentos que tornam o sangue mais fluido (cumarina, varfarina),
- certos medicamentos antivirais (sorivudina e brivudina),
- medicamentos para tratar convulsões ou tremor (fenitoína),
- interferon alfa,
- radioterapia e alguns medicamentos usados para tratar o câncer (ácido folínico, oxaliplatina, bevacizumab, cisplatina, irinotecano),
- medicamentos usados para tratar deficiências de ácido fólico.
Xeloda com comida e bebida
Deve tomar o Xeloda 30 minutos após o final da refeição.
Avisos É importante saber que:
Gravidez e amamentação
Antes de iniciar o tratamento, informe o seu médico se estiver grávida, suspeitar ou planejar engravidar. Não deve tomar Xeloda se estiver grávida ou com suspeita de gravidez. Não deve amamentar enquanto está a tomar Xeloda. Consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Condução e utilização de máquinas
Xeloda pode fazer você se sentir tonto, enjoado ou cansado. Por conseguinte, é possível que Xeloda possa afetar a capacidade de conduzir ou utilizar máquinas.
Xeloda contém lactose anidra
Se foi informado pelo seu médico que tem “intolerância a alguns açúcares, contacte-o antes de tomar este medicamento.
Dose, Método e Tempo de Administração Como usar o Xeloda: Posologia
Tome este medicamento sempre de acordo com as indicações do seu médico ou farmacêutico. Se você não tiver certeza, consulte o seu médico ou farmacêutico.
O Xeloda só deve ser prescrito por um médico especialista na utilização de medicamentos antineoplásicos.
Os comprimidos de Xeloda devem ser engolidos inteiros com água 30 minutos após o final da refeição.
O seu médico irá prescrever a dosagem e o regime de tratamento adequados para si. A dosagem de Xeloda é baseada na área de superfície corporal. Isso é calculado a partir da altura e do peso. A dose usual para adultos é de 1250 mg / m2 de área de superfície corporal duas vezes ao dia (manhã e noite). Dois exemplos são propostos: uma pessoa com peso de 64 kg e altura de 1,64 m tem uma área de superfície corporal de 1,7 m2 e deve tomar 4 comprimidos de 500 mg e 1 comprimido de 150 mg duas vezes ao dia. Uma pessoa com peso de 80 kg e altura de 1,80 m tem área de superfície corporal de 2,00 m2 e deve tome 5 comprimidos de 500 mg duas vezes ao dia.
Os comprimidos de Xeloda são geralmente tomados durante 14 dias, seguidos de um período de descanso de 7 dias (durante o qual nenhum comprimido é tomado). Esses 21 dias correspondem a um ciclo de terapia.
Em combinação com outros medicamentos, a dose usual para adultos pode ser inferior a 1250 mg / m2 de área de superfície corporal e os comprimidos podem precisar ser tomados por um período de tempo diferente (por exemplo, todos os dias, sem qualquer período de descanso).
O seu médico irá dizer-lhe qual a dose que deve tomar, quando deve tomá-la e quanto tempo deve tomá-la.
O seu médico pode prescrever uma combinação de comprimidos de 150 mg e 500 mg para cada dosagem.
- Tome os comprimidos de manhã e à noite, conforme prescrito pelo seu médico.
- Tome os comprimidos 30 minutos após terminar a refeição (pequeno-almoço e jantar).
- É importante tomar todos os medicamentos prescritos pelo seu médico.
Sobredosagem O que fazer se tiver tomado Xeloda em demasia
Se você tomar mais Xeloda do que deveria
Se tomar mais Xeloda do que deveria, contacte o seu médico assim que possível antes de tomar a próxima dose.
Se tomar mais Xeloda do que deveria, pode sentir os seguintes efeitos secundários: náuseas ou vómitos, diarreia, inflamação ou ulceração do intestino ou da boca, dor ou hemorragia intestinal ou estômago, ou depressão da medula óssea (redução de alguns tipo de células sanguíneas). Se tiver algum destes sintomas, contacte o seu médico imediatamente.
Se você se esquecer de tomar Xeloda:
Não tome a dose esquecida e não duplique a próxima. Em vez disso, continue com a sua dosagem normal e contacte o seu médico.
Se você parar de tomar Xeloda:
Parar o tratamento com capecitabina não causa efeitos secundários.Parar com capecitabina, se estiver a tomar anticoagulantes cumarínicos (contendo por exemplo fenprocumom), pode exigir que o seu médico altere a dose do anticoagulante.
Caso ainda tenha dúvidas sobre a utilização deste produto, fale com o seu médico ou farmacêutico.
Efeitos colaterais Quais são os efeitos colaterais do Xeloda
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
PARE de tomar Xeloda imediatamente e contacte o seu médico se algum dos seguintes sintomas se desenvolver:
- Diarreia: se tiver um aumento de 4 ou mais evacuações por dia em comparação com as evacuações normais ou diarreia nocturna.
- Vômito: se vomitar mais de uma vez em um período de 24 horas.
- Náusea: se você perder o apetite e a quantidade de comida ingerida por dia for muito menor do que o normal.
- Estomatite: se tiver dor, vermelhidão, inchaço ou úlceras na boca ou na garganta.
- Reação cutânea mão-pé: se tiver dor, inchaço e vermelhidão ou formigamento nas mãos e / ou pés.
- Febre: se tem uma temperatura corporal de 38 ° C ou superior.
- Infecção: se tiver algum sinal de infecção por bactérias ou vírus ou outros organismos.
- Dor no peito: se sentir dor localizada no centro do peito, especialmente se ocorrer durante o exercício.
- Síndrome de Steven-Johnson: se você tiver uma erupção cutânea dolorosa vermelha ou roxa que se espalha e forma bolhas e / ou outras lesões que começam a aparecer na membrana mucosa (por exemplo, boca e lábios), especialmente se você já teve sensibilidade à luz, infecções de o sistema respiratório (por exemplo, bronquite) e / ou febre.
Se detectados precocemente, esses efeitos colaterais geralmente melhoram 2-3 dias após a interrupção do medicamento. Se os sintomas persistirem, entre em contato com seu médico imediatamente. O seu médico pode aconselhá-lo a retomar o tratamento com uma dose mais baixa do medicamento.
Além dos listados acima, outros efeitos colaterais muito comuns relatados com o uso de Xeloda sozinho, que podem afetar mais de 1 em cada 10 pessoas, são:
- dor abdominal
- erupção cutânea, pele seca ou coceira
- cansaço
- perda de apetite (anorexia).
Esses efeitos colaterais podem se tornar graves. Portanto, sempre contate seu médico imediatamente quando notar um efeito colateral. O seu médico irá dizer-lhe para diminuir a dose e / ou interromper temporariamente o tratamento com Xeloda. Isso ajudará a reduzir a probabilidade de o efeito colateral persistir ou transformá-lo em um efeito colateral sério.
Outros efeitos colaterais são:
Os efeitos colaterais comuns (podem afetar até 1 em cada 10 pessoas) incluem:
- diminuição do número de glóbulos brancos ou vermelhos no sangue (observada em testes),
- desidratação, perda de peso,
- falta de sono (insônia), depressão,
- dor de cabeça, sonolência, tontura, sensação anormal na pele (dormência ou formigamento), mudança no paladar,
- irritação nos olhos, aumento de lacrimejamento, olhos vermelhos (conjuntivite),
- inflamação das veias (tromboflebite),
- falta de ar, sangramento nasal, tosse, coriza,
- herpes labial ou outras infecções por herpes,
- infecções dos pulmões ou do sistema respiratório (por exemplo, pneumonia ou bronquite),
- sangramento intestinal, constipação, dor abdominal superior, indigestão, excesso de ar, boca seca,
- erupção cutânea, queda de cabelo (alopecia), vermelhidão da pele, pele seca, coceira, descoloração da pele, perda de pele, inflamação da pele, alterações nas unhas,
- dor nas articulações ou membros (extremidades), tórax ou costas,
- febre, inchaço dos membros, mal-estar,
- problemas da função hepática (observados em análises ao sangue) e aumento da bilirrubina no sangue (excretada pelo fígado).
Os efeitos colaterais incomuns (podem afetar menos de 1 em 100 pessoas) incluem:
- infecção do sangue, infecção do trato urinário, infecção da pele, infecção do nariz e garganta, infecções fúngicas (incluindo as da boca), gripe, gastroenterite, abscesso,
- inchaços suaves sob a pele (lipoma),
- diminuição das células sanguíneas, incluindo plaquetas, afinamento do sangue (observado em testes),
- alergia,
- diabetes, diminuição do potássio no sangue, desnutrição, aumento dos triglicerídeos no sangue,
- estado confusional, ataques de pânico, depressão do humor, diminuição da libido,
- dificuldade de falar, comprometimento da memória, perda de coordenação do movimento, distúrbio do equilíbrio, desmaios, danos nos nervos (neuropatia) e problemas com os sentidos,
- visão turva ou dupla,
- tontura, dor de ouvido,
- batimento cardíaco irregular e palpitações (arritmia), dor no peito e ataque cardíaco (ataque cardíaco),
- coágulos sanguíneos nas veias profundas, pressão alta ou baixa, rubor, frio nos membros (extremidades), manchas roxas na pele,
- coágulos de sangue nas veias do pulmão (embolia pulmonar), colapso do pulmão, perda de sangue com tosse, asma, falta de ar com o esforço,
- obstrução intestinal, acúmulo de líquido no abdômen, inflamação do intestino delgado ou grosso, estômago ou esôfago, dor na parte inferior do abdômen, desconforto abdominal, azia (refluxo de comida do estômago), sangue nas fezes,
- icterícia (amarelecimento da pele e olhos),
- úlcera e bolhas na pele, reações cutâneas à luz solar, vermelhidão das palmas das mãos, inchaço ou dor no rosto,
- inchaço ou rigidez das articulações, dor nos ossos, fraqueza ou rigidez muscular,
- coleção de fluidos nos rins, aumento da frequência urinária à noite, incontinência, sangue na urina, aumento da creatinina no sangue (sinal de disfunção renal),
- sangramento incomum da vagina,
- inchaço (edema), calafrios e rigidez.
Alguns desses efeitos colaterais são comuns quando a capecitabina é usada com outros medicamentos para tratar o câncer. Outros efeitos colaterais observados neste contexto são:
Os efeitos colaterais comuns (podem afetar até 1 em cada 10 pessoas) incluem:
- diminuição do sódio, magnésio e cálcio no sangue, aumento do açúcar no sangue,
- dor no nervo,
- zumbido nos ouvidos (zumbido), perda de audição,
- inflamação das veias,
- soluços, voz alterada,
- dor ou sensação alterada / anormal na boca, dor na mandíbula,
- suores, suores noturnos,
- espasmo muscular,
- dificuldade para urinar, sangue ou proteína na urina,
- hematomas ou reações no local da injeção (causadas por medicamentos administrados por injeção ao mesmo tempo).
Efeitos colaterais raros (podem afetar até 1 em 1.000 pessoas) incluem:
- estreitamento ou bloqueio do canal lacrimal (estenose do canal lacrimal),
- insuficiência hepática,
- inflamação que leva à disfunção ou bloqueio da secreção biliar (hepatite colestática),
- mudanças específicas no eletrocardiograma (prolongamento QT),
- certos tipos de arritmias (incluindo fibrilação ventricular, torsades de pointes e bradicardia),
- inflamação dos olhos causando dor e potenciais problemas de visão,
- inflamação da pele causando manchas vermelhas e descamação devido a uma doença do sistema imunológico.
Efeitos colaterais muito raros (podem afetar até 1 em 10.000 pessoas) incluem:
- reações cutâneas graves, como erupção na pele, ulceração e bolhas, que podem envolver úlceras da boca, nariz, órgãos genitais, mãos, pés e olhos (olhos vermelhos e inchados).
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico, farmacêutico ou enfermeiro. Isto inclui quaisquer efeitos secundários possíveis não listados neste folheto. Também pode comunicar os efeitos secundários diretamente através do sistema nacional de notificação listado no Apêndice V. efeitos secundários pode ajudar fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
Manter fora da vista e do alcance das crianças.
Não armazene em temperaturas acima de 30 ° C.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no rótulo após “VAL.” O prazo de validade corresponde ao último dia do mês.
Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Composição e forma farmacêutica
O que Xeloda contém
A substância ativa é a capecitabina (150 mg para cada comprimido revestido por película).
Os outros excipientes são:
- Núcleo do comprimido: lactose anidra, croscarmelose de sódio, hipromelose, celulose microcristalina, estearato de magnésio.
- Revestimento do comprimido: hipromelose, dióxido de titânio (E171), óxido de ferro amarelo e vermelho (E172), talco.
Qual a aparência de Xeloda e conteúdo da embalagem
Comprimido revestido por película, pêssego claro, biconvexo, oblongo, com a gravação "150" numa das faces e "Xeloda" na outra.
A embalagem de Xeloda 150 mg comprimido revestido por película contém 60 comprimidos revestidos por película (6 blisters de 10 comprimidos).
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
COMPRIMIDOS DE XELODA 150 MG REVESTIDOS COM PELÍCULA
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Cada comprimido revestido por película contém 150 mg de capecitabina.
Excipiente (s) com efeito conhecido:
cada comprimido revestido por película contém 15,6 mg de lactose anidra.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Comprimido revestido por película.
Os comprimidos revestidos por película de Xeloda 150 mg são pêssegos claros, biconvexos, oblongos, gravados com “150” numa das faces e “Xeloda” na outra.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
Xeloda está indicado como terapêutica adjuvante em doentes submetidos a cirurgia para cancro do cólon em estádio III (Dukes C) (ver secção 5.1).
Xeloda está indicado para o tratamento do cancro colo-rectal metastático (ver secção 5.1).
Xeloda é indicado para o tratamento de primeira linha do cancro gástrico avançado em combinação com um regime à base de platina (ver secção 5.1).
Xeloda em combinação com docetaxel (ver secção 5.1) está indicado para o tratamento de doentes com cancro da mama localmente avançado ou metastático após falha da quimioterapia citotóxica. A terapia anterior deve ter incluído uma "antraciclina. Além disso, Xeloda é indicado como monoterapia para o tratamento de pacientes com câncer de mama localmente avançado ou metastático após falha de um taxano e um regime de quimioterapia contendo antraciclina ou para os quais uma antraciclina não é indicada." terapia adicional com antraciclina.
04.2 Posologia e método de administração
O Xeloda só deve ser prescrito por um médico especialista na utilização de medicamentos antineoplásicos. Recomenda-se uma monitorização cuidadosa para todos os doentes durante o primeiro ciclo de tratamento.
O tratamento deve ser interrompido se ocorrer toxicidade grave ou progressão da doença. Os cálculos de dose padrão e reduzida com base na área de superfície corporal para dosagens iniciais de Xeloda de 1250 mg / m2 e 1000 mg / m2 estão detalhados nas Tabelas 1 e 2, respectivamente.
Dosagem
Posologia recomendada (ver seção 5.1):
Monoterapia
Câncer de cólon, colorretal e mama
No tratamento em monoterapia, a dose inicial recomendada de capecitabina no tratamento adjuvante do cólon, câncer colorretal metastático ou câncer de mama localmente avançado ou metastático é de 1250 mg / m2, administrado duas vezes ao dia (manhã e à noite; total diário de 2500 mg / m2) durante 14 dias, seguido de um período de descanso de 7 dias. A terapia adjuvante em pacientes com câncer de cólon em estágio III é recomendada por um total de 6 meses.
Terapia de associação
Câncer de cólon, colorretal e gástrico
No tratamento de combinação, a dose inicial recomendada de capecitabina deve ser reduzida para 800 - 1000 mg / m2 quando administrada duas vezes ao dia por 14 dias, seguida por um período de descanso de 7 dias ou para 625 mg / m2 duas vezes ao dia. Quando administrada continuamente (ver seção 5.1). Em combinação com irinotecano, a dose inicial recomendada é de 800 mg / m2 quando administrada duas vezes ao dia por 14 dias, seguida por um período de descanso de 7 dias em combinação com irinotecano 200 mg / m2 no dia 1. A introdução de bevacizumabe no regime de combinação não tem efeito sobre a dose inicial de capecitabina. Em pacientes tratados com a combinação capecitabina mais cisplatina, a pré-medicação para manter a hidratação adequada e o tratamento antiemético devem ser iniciados antes da administração de cisplatina, de acordo com o Resumo das Características do Medicamento da cisplatina. Pré-medicação com antieméticos. é recomendado em pacientes tratados com a combinação de capecitabina com oxaliplatina, de acordo com o Resumo das Características do Medicamento da oxaliplatina. Uma duração de 6 meses de tratamento adjuvante é recomendada em pacientes com câncer de cólon em estágio III.
Câncer de mama
Em combinação com docetaxel, a dose inicial recomendada de capecitabina no tratamento do câncer de mama metastático é 1250 mg / m2 duas vezes ao dia por 14 dias, seguido por um período de descanso de 7 dias, em combinação com docetaxel 75 mg / m2 em 1 hora intravenosa infusão a cada 3 semanas. Em pacientes recebendo a combinação de capecitabina e docetaxel, a pré-medicação com um corticosteroide oral, como a dexametasona, deve ser iniciada antes da administração do docetaxel, de acordo com o Resumo das Características do Medicamento do docetaxel.
Cálculo da dose de Xeloda
Tabela 1 Cálculos de dose padrão e reduzida de capecitabina com base na área de superfície corporal, dose inicial de 1250 mg / m2
Tabela 2 Cálculos de dose padrão e reduzida de capecitabina com base na área de superfície corporal, dose inicial de 1000 mg / m2
Ajustes de dosagem durante o tratamento:
Em geral
A toxicidade causada pela administração de capecitabina pode ser controlada com tratamento sintomático e / ou modificação da dose (interrupção do tratamento ou redução da dose). Uma vez que a dose tenha sido reduzida, não deve ser aumentada posteriormente. No caso de toxicidades que, na opinião do médico assistente, são improváveis de se tornarem graves ou fatais, como alopecia, alteração do paladar, alterações das unhas, o tratamento pode ser continuado com a mesma dose sem redução ou interrupção. Os doentes a tomar capecitabina devem ser avisados da necessidade de interromper o tratamento imediatamente se ocorrer toxicidade moderada ou grave. As doses de capecitabina excluídas devido à toxicidade não podem ser substituídas. A seguir estão as modificações de dose recomendadas em caso de toxicidade:
Tabela 3 Cronograma de redução da dose de capecitabina (ciclo de 3 semanas ou tratamento contínuo)
* De acordo com os Critérios Comuns de Toxicidade (versão 1) do Grupo de Ensaios Clínicos do Instituto Nacional do Câncer do Canadá (NCIC CGT) ou os Critérios Comuns de Terminologia para Eventos Adversos (CTCAE) do Programa de Avaliação de Terapia do Câncer, Instituto Nacional do Câncer dos EUA, versão 4.0 . Para síndrome mão-pé e hiperbilirrubinemia, ver secção 4.4.
Hematologia
Pacientes com contagens basais de neutrófilos
Modificações de dose para toxicidade quando a capecitabina é usada em um ciclo de 3 semanas em combinação com outros medicamentos
Quando a capecitabina é usada em ciclos de 3 semanas em combinação com outros medicamentos, as modificações da dose para toxicidade devem ser realizadas de acordo com a tabela 3 acima para a capecitabina e de acordo com o Resumo das Características do Medicamento relevante para o (s) outro (s) medicamento (s). / O .
No início do tratamento, se o adiamento do tratamento for indicado para a capecitabina ou para o (s) outro (s) medicamento (s), a administração de todos os medicamentos deve ser adiada até o momento necessário para retomar a administração de todos os medicamentos.
Durante o tratamento, para as toxicidades consideradas pelo médico assistente como não relacionadas com a capecitabina, o tratamento com capecitabina deve ser continuado e a dose do outro medicamento ajustada de acordo com a informação de prescrição relevante.
Se o (s) outro (s) medicamento (s) tiver (m) de ser descontinuado (s) definitivamente, o tratamento com capecitabina pode ser retomado assim que forem cumpridos os requisitos para a reintrodução de capecitabina.
Esta abordagem se aplica a todas as indicações e todas as populações especiais de pacientes.
Modificações da dose para toxicidade quando a capecitabina é usada como tratamento contínuo em combinação com outros medicamentos
As modificações da dose para toxicidade quando a capecitabina é usada como tratamento contínuo em combinação com outros medicamentos devem ser realizadas de acordo com a Tabela 3 acima para a capecitabina e de acordo com o Resumo das Características do Medicamento relevante para o (s) outro (s) medicamento (s).
Ajustes de dosagem em populações específicas de pacientes:
Função hepática prejudicada
Os dados de segurança e eficácia são insuficientes para fornecer orientação sobre os ajustes de dose em pacientes com insuficiência hepática. Não existem dados sobre a insuficiência hepática devida a cirrose ou hepatite.
Função renal prejudicada
Capecitabina é contra-indicada em pacientes com insuficiência renal grave (depuração da creatinina inferior a 30 ml / min [Cockcroft e Gault] no início do estudo). A incidência de reações adversas de grau 3 ou 4 em pacientes com insuficiência renal moderada (depuração da creatinina 30-50 mL / min na linha de base) é maior do que na população total. Recomenda-se uma redução de 75% para uma dose inicial de 1250 mg / m2 em pacientes com insuficiência renal moderada na linha de base. Não é necessária redução da dose para uma dose inicial de 1000 mg / m2 em pacientes com insuficiência renal moderada na linha de base. dose inicial em pacientes com insuficiência renal leve (depuração da creatinina 51-80 ml / min em linha de base). Se o paciente desenvolver um evento adverso de grau 2, 3 ou 4 durante o tratamento, monitoramento cuidadoso e "interrupção imediata do tratamento e a próxima dose devem ser ajustadas conforme indicado na Tabela 3 acima. Se a depuração de creatinina calculada cair durante o tratamento nde abaixo de 30 ml / min, Xeloda deve ser descontinuado. Estas recomendações sobre ajustes de dose no compromisso renal aplicam-se tanto à monoterapia quanto ao uso de combinação (ver também a seção “Idosos” abaixo).
Cidadãos idosos
Não é necessário ajuste da dose inicial quando se usa capecitabina isoladamente. No entanto, pacientes ≥ 60 anos de idade em comparação com indivíduos mais jovens relataram reações adversas relacionadas ao tratamento de Grau 3 ou 4 com maior frequência.
Quando a capecitabina foi utilizada em combinação com outros medicamentos, os doentes idosos (≥ 65 anos) tiveram mais reacções adversas de grau 3 e 4, incluindo aquelas que conduziram à descontinuação do tratamento, do que os doentes mais jovens. É aconselhável monitorização cuidadosa dos doentes ≥ 60 anos de idade.
- Em combinação com docetaxel: Foi observado um aumento da incidência de reações adversas relacionadas com o tratamento de grau 3 ou 4 e reações adversas graves relacionadas com o tratamento em doentes com 60 ou mais anos de idade (ver secção 5.1) .Uma posologia inicial de capecitabina reduzida para 75% (950 mg / m2 duas vezes ao dia) em pacientes com 60 anos de idade ou mais. Se não ocorrer toxicidade em pacientes ≥ 60 anos de idade tratados com uma dose inicial reduzida de capecitabina em combinação com docetaxel, a dose de capecitabina pode ser aumentada com cautela para 1250 mg / m2 duas vezes Diário.
População pediátrica
Não existe utilização relevante de capecitabina na população pediátrica nas indicações de cancro do cólon, colo-rectal, gástrico e da mama.
Método de administração
Os comprimidos de Xeloda devem ser engolidos com água 30 minutos após o final da refeição.
04.3 Contra-indicações
• História de reações graves ou inesperadas à terapia com fluoropirimidina.
• Hipersensibilidade à capecitabina ou a qualquer um dos excipientes listados na seção 6.1 ou ao fluorouracil.
• Em doentes com ausência completa conhecida de atividade da dihidropirimidina desidrogenase (DPD) (ver secção 4.4).
• Durante a gravidez e amamentação.
• Em pacientes com formas graves de leucopenia, neutropenia ou trombocitopenia.
• Em pacientes com insuficiência hepática grave.
• Em pacientes com insuficiência renal grave (depuração da creatinina inferior a 30 ml / min).
• Durante o tratamento com sorivudina ou seus análogos quimicamente relacionados, como a brivudina (ver secção 4.5).
• Se houver contra-indicações para qualquer um dos medicamentos do regime de combinação, esse medicamento não deve ser usado.
04.4 Advertências especiais e precauções adequadas de uso
o toxicidades limitantes de dose incluem diarreia, dor abdominal, náusea, estomatite e síndrome mão-pé (reação cutânea mão-pé, eritrodisestesia palmo-plantar). A maioria das reações adversas é reversível e não requer a descontinuação permanente da terapia, embora a interrupção ou redução da dose possa ser necessária.
Diarréia. Pacientes com diarreia grave devem ser monitorados de perto e, em caso de desidratação, devem ser administrados líquidos e eletrólitos. Podem ser administrados tratamentos antidiarreicos padrão (por exemplo, loperamida). Diarreia de grau 2 de acordo com os Critérios de Toxicidade Comuns do NCIC significa um aumento de 4 para 6 descargas por dia ou descargas noturnas, para diarreia de grau 3 um aumento de 7 a 9 descargas por dia ou incontinência e má absorção, e para diarreia de Grau 4 e aumento de ≥10 altas por dia ou diarreia com forte sangramento ou necessidade de suporte parenteral. Se necessário, deve ser feita uma redução da dose (ver secção 4.2).
Desidratação. A desidratação deve ser evitada ou corrigida quando ocorrer. Pacientes com anorexia, astenia, náusea, vômito ou diarreia podem ficar rapidamente desidratados. A desidratação pode causar insuficiência renal aguda, especialmente em pacientes com insuficiência renal preexistente ou quando a capecitabina é administrada em combinação com drogas nefrotóxicas conhecidas. A insuficiência renal aguda secundária à desidratação pode ser potencialmente fatal. Se ocorrer desidratação de grau 2 (ou superior), o tratamento com capecitabina deve ser interrompido imediatamente e a desidratação corrigida. O tratamento não deve ser retomado até que o paciente tenha sido reidratado e qualquer causa precipitante corrigida ou controlada. Devem ser feitas modificações da dose para o evento adverso precipitante, conforme necessário (ver seção 4.2).
Síndrome mão-pé (também conhecido como reação cutânea mão-pé ou eritrodisestesia palmo-plantar ou eritema das extremidades induzido por quimioterapia). A síndrome mão-pé de grau 1 é definida como dormência, disestesia / parestesia, formigamento, edema ou eritema indolor das mãos e / ou pés e / ou desconforto que não impede as atividades normais do paciente.
A síndrome mão-pé de grau 2 é definida como eritema doloroso e edema nas mãos e / ou pés e / ou desconforto que afeta as atividades diárias do paciente.
A síndrome mão-pé de grau 3 é definida como descamação úmida, ulceração, bolhas e dor intensa nas mãos e / ou pés e / ou desconforto severo que torna impossível para o paciente trabalhar ou realizar atividades diárias. Se ocorrer síndrome do pé, suspenda a administração de capecitabina até que a intensidade dos sintomas seja resolvida ou reduzida para Grau 1. Após o início da síndrome mão-pé de Grau 3, as doses subsequentes de capecitabina devem ser reduzidas. Quando capecitabina e cisplatina são usadas em combinação, o uso de vitamina B6 (piridoxina) para tratamento profilático sintomático ou secundário da síndrome mão-pé não é recomendado, pois casos publicados mostraram que pode reduzir a eficácia da cisplatina. Existem algumas evidências de que o dexpantenol é eficaz na profilaxia da síndrome mão-pé em pacientes tratados com Xeloda.
Cardiotoxicidade. A terapia com fluoropirimidina foi associada a cardiotoxicidade, incluindo infarto do miocárdio, angina, arritmia, choque cardiogênico, morte súbita e alterações eletrocardiográficas (incluindo casos muito raros de prolongamento do intervalo QT). Essas reações adversas podem ocorrer mais comumente em pacientes com história prévia de artéria coronária Foram notificados casos de arritmia cardíaca (incluindo fibrilhação ventricular, torsades de pointes e bradicardia), angina de peito, enfarte do miocárdio, insuficiência cardíaca e cardiomiopatia em doentes a tomar capecitabina. angina de peito significativa deve ter-se especial cuidado (ver secção 4.8).
Hipo ou hipercalcemia. Foram notificados casos de hipo ou hipercalcemia durante o tratamento com capecitabina. Deve ter-se cuidado especial em doentes com história pré-existente de hipo ou hipercalcemia (ver secção 4.8).
Doenças do sistema nervoso central ou periférico. Os doentes com doenças do sistema nervoso central ou periférico, por exemplo metástases cerebrais ou neuropatia, devem ser vistos com cuidado (ver secção 4.8).
Diabetes mellitus ou distúrbios eletrolíticos. Pacientes com diabetes mellitus ou distúrbios eletrolíticos, dada a possibilidade de agravamento durante o tratamento com capecitabina, devem ser considerados com cautela.
Anticoagulantes derivados da cumarina. Num estudo de interação com a administração de uma dose única de varfarina, houve um aumento significativo na AUC média (+ 57%) da S-varfarina. Esses dados sugerem uma "interação, provavelmente devido à" inibição da isoenzima 2C9 do citocromo P450 pela capecitabina. Pacientes tomando anticoagulantes orais derivados de cumarina juntamente com capecitabina devem ser monitorados regularmente para a possível ocorrência de alterações nos parâmetros de coagulação (INR ou protrombina tempo) e a dose de anticoagulantes deve ser ajustada em conformidade (ver secção 4.5).
Função hepática prejudicada. Na ausência de dados de segurança e eficácia em pacientes com função hepática comprometida, o uso de capecitabina deve ser monitorado de perto em pacientes com disfunção hepática leve a moderada, independentemente da presença ou ausência de metástases hepáticas. A administração de capecitabina deve ser interrompida se ocorrem elevações relacionadas ao tratamento na bilirrubina maiores que 3,0 x ULN ou elevações relacionadas ao tratamento nas aminotransferases hepáticas (ALT, AST) maiores que 2,5 x LSN. A monoterapia pode ser retomada quando a bilirrubina diminui para ≤3,0 x ULN ou diminuem as aminotransferases hepáticas para ≤ 2,5 x ULN.
Função renal prejudicada. A incidência de reações adversas de grau 3 ou 4 em doentes com compromisso renal moderado (depuração da creatinina 30-50 ml / min) é mais elevada do que na população geral (ver secções 4.2 e 4.3).
Deficiência de dihidropirimidina desidrogenase (DPD): Toxicidade rara, inesperada e grave (por exemplo, estomatite, diarreia, mucosite, neutropenia e neurotoxicidade) associada ao 5-FU foi relacionada a um déficit na atividade de DPD.
Pacientes com baixa ou nenhuma atividade de DPD, uma enzima envolvida na degradação do fluorouracil, apresentam risco aumentado de reações adversas graves, fatais ou fatais causadas pelo fluorouracil. Embora a deficiência de DPD não possa ser identificada com precisão, sabe-se que os pacientes com certas mutações homozigóticas ou heterozigóticas compostas do locus do gene DPYD, que causam ausência completa ou quase completa da atividade enzimática DPD (conforme determinado por análise laboratorial), apresentam o maior risco de toxicidade com risco de vida ou fatal e não devem ser tratados com Xeloda (ver secção 4.3). Nenhuma dose foi considerada segura para pacientes com ausência total de atividade DPD.
Pacientes com deficiência parcial de DPD (como aqueles com mutações heterozigotas no DPYD) e para os quais o benefício de Xeloda é considerado compensar os seus riscos (tendo em consideração a adequação de um regime alternativo de quimioterapia sem fluopirimidina) deve ser tratado com extrema precaução e frequentemente monitorizado com ajuste da dose de acordo com a toxicidade. Não existem dados suficientes para recomendar uma dose específica em pacientes com atividade DPD parcial medida por um teste específico.
Toxicidades potencialmente fatais, como episódios agudos de sobredosagem, podem ocorrer em doentes com deficiência de DPD não identificada que são tratados com capecitabina (ver secção 4.9). Em caso de toxicidade aguda de grau 2-4, o tratamento deve ser interrompido imediatamente. A descontinuação permanente do tratamento deve ser considerada com base na avaliação clínica do início, duração e gravidade das toxicidades observadas.
Complicações oftalmológicas: Os pacientes devem ser monitorados de perto quanto a complicações oftalmológicas, como ceratite e distúrbios da córnea, especialmente se tiverem histórico anterior de distúrbios oculares. O tratamento das doenças oculares deve ser iniciado de forma clinicamente apropriada.
Reações cutâneas graves: Xeloda pode induzir reações cutâneas graves, como a síndrome de Stevens-Johnson e necrólise epidérmica tóxica. Em doentes que apresentem uma reação cutânea grave durante o tratamento com Xeloda, este medicamento deve ser descontinuado definitivamente.
Uma vez que este medicamento contém lactose anidra como excipiente, os doentes com formas hereditárias raras de intolerância à galactose, deficiência da enzima lactase Lapp e má absorção de glucose-galactose não devem tomar este medicamento.
04.5 Interações com outros medicamentos e outras formas de interação
Os estudos de interação foram realizados apenas em adultos.
Interações com outros medicamentos:
Substratos do citocromo P-450 2C9: Além dos estudos com varfarina, não foram realizados estudos formais de interação medicamentosa entre a capecitabina e outros substratos do CYP2C9. Deve-se ter cuidado ao administrar capecitabina junto com substratos 2C9 (por exemplo, fenitoína). Ver também interação com outros anticoagulantes derivados da cumarina e seção 4.4.
Anticoagulantes derivados de cumarinaAlterações nos parâmetros de coagulação e / ou hemorragias foram relatadas em pacientes tratados concomitantemente com capecitabina e anticoagulantes derivados de cumarina, como varfarina e fenprocumon. Estas reações ocorreram durante um período de alguns dias a vários meses após o início da terapia com capecitabina e, em alguns casos, dentro de um mês após a interrupção da terapia com capecitabina. Num estudo clínico de interação farmacocinética, após a administração de uma dose única de 20 mg de varfarina, o tratamento com capecitabina aumentou a AUC da S-varfarina em 57% com um aumento de 91% no INR. Uma vez que o metabolismo da R-varfarina não foi alterado, esses dados sugerem que a capecitabina reduz a isoenzima 2C9, mas não tem efeito sobre as isoenzimas 1A2 e 3A4. Pacientes tomando anticoagulantes derivados da cumarina concomitantemente com capecitabina devem ser monitorados regularmente para a possível ocorrência de alterações em os parâmetros de coagulação (PT ou INR) e a dosagem dos anticoagulantes devem ser ajustados em conformidade.
Fenitoína: Aumentos nas concentrações plasmáticas de fenitoína foram registrados durante o uso concomitante de capecitabina e fenitoína, resultando em sintomas de intoxicação por fenitoína em casos individuais Os pacientes que tomam fenitoína concomitantemente com capecitabina devem ser monitorados regularmente para qualquer ocorrência de aumento das concentrações plasmáticas de fenitoína.
Ácido folínico / ácido fólico: Um estudo envolvendo a combinação de capecitabina e ácido folínico mostrou que o ácido folínico não tem efeito significativo na farmacocinética da capecitabina e seus metabólitos. No entanto, o ácido folínico produz efeitos na farmacodinâmica da capecitabina, cuja toxicidade pode ser aumentada pelo ácido folínico: a dose máxima tolerada (DMT) de monoterapia com capecitabina em regimes intermitentes é de 3000 mg / m2 por dia, enquanto quando a capecitabina foi associada ao ácido folínico ( 30 mg po duas vezes ao dia), a dose máxima tolerada caiu para apenas 2.000 mg / m2 por dia. O aumento da toxicidade pode ser relevante quando se muda de 5-FU / LV para um regime baseado em capecitabina. Devido à semelhança entre o ácido folínico e o ácido fólico, o aumento da toxicidade também pode ser relevante com a suplementação de ácido fólico no tratamento da deficiência de folato .
Sorivudina e análogos: Foi relatada uma interação medicamentosa clinicamente significativa entre a sorivudina e o 5-FU, resultante da inibição da dihidropirimidina desidrogenase pela sorivudina. Essa interação, que leva ao aumento da toxicidade da fluoropirimidina, é potencialmente fatal. Por este motivo, a capecitabina não deve ser administrada concomitantemente com sorivudina ou seus análogos quimicamente relacionados, como a brivudina (ver secção 4.3). Deve ser observado um período de descanso de pelo menos 4 semanas entre o final do tratamento com sorivudina ou seus análogos quimicamente relacionados, como a brivudina, e o início da terapia com capecitabina.
Antiácidos: Foi investigado o efeito de um antiácido contendo hidróxido de alumínio e hidróxido de magnésio na farmacocinética da capecitabina.Houve um ligeiro aumento nas concentrações plasmáticas de capecitabina e um metabólito (5 "-DFCR); não houve efeito nos 3 metabólitos principais (5 "-DFUR, 5-FU e FBAL).
Alopurinol: Foram observadas interações de 5-FU com alopurinol, com possível diminuição da eficácia de 5-FU. O uso concomitante de alopurinol e capecitabina deve ser evitado.
Interferon alfa: a dose máxima tolerada (MTD) de capecitabina foi de 2.000 mg / m2 por dia quando administrada em combinação com interferão alfa-2a (3 MUI / m2 por dia), em comparação com 3.000 mg / m2 por dia quando a capecitabina foi administrada isoladamente.
Radioterapia: A dose máxima tolerada (MTD) de monoterapia de capecitabina usando o regime intermitente é de 3000 mg / m2 por dia, enquanto, quando combinada com radioterapia para câncer retal, a dose máxima tolerada (MTD) de capecitabina é de 2000 mg / m2 por dia, usando ou uma dosagem contínua ou uma dosagem diária de segunda a sexta-feira em conjunto com o ciclo de tratamento de radioterapia de 6 semanas.
Oxaliplatina: Não houve diferença clinicamente significativa na exposição à capecitabina ou seus metabólitos, platina livre ou platina total quando a capecitabina foi administrada em combinação com oxaliplatina ou em combinação com oxaliplatina e bevacizumab.
Bevacizumab: Não houve efeito clinicamente significativo do bevacizumab nos parâmetros farmacocinéticos da capecitabina ou seus metabólitos na presença de oxaliplatina.
Interação com comida
Em todos os estudos clínicos, os pacientes foram aconselhados a tomar capecitabina 30 minutos após uma refeição. Uma vez que os dados atuais de segurança e eficácia se baseiam na administração do medicamento com alimentos, recomenda-se que a capecitabina seja administrada com alimentos.A administração com alimentos diminui a taxa de absorção da capecitabina (ver secção 5.2).
04.6 Gravidez e lactação
Mulheres em idade fértil / contracepção em homens e mulheres
As mulheres com potencial para engravidar devem ser aconselhadas a evitar o risco de gravidez durante o tratamento com capecitabina. Se ocorrer gravidez durante o tratamento com capecitabina, a paciente deve ser informada do risco potencial para o feto. Deve ser utilizado um método contraceptivo eficaz durante o tratamento.
Gravidez
Não foram realizados estudos com capecitabina em mulheres grávidas; no entanto, pode-se presumir que a capecitabina, quando administrada a mulheres grávidas, pode causar danos ao feto. Em estudos de toxicidade reprodutiva em animais, a administração de capecitabina resultou em letalidade embrionária e teratogenicidade. Estes resultados são os efeitos esperados dos derivados de fluoropirimidina. Capecitabina é contra-indicada na gravidez.
Hora da alimentação
Não se sabe se a capecitabina é excretada no leite humano. Quantidades significativas de capecitabina e seus metabólitos foram encontradas no leite de camundongo em lactação.A amamentação deve ser interrompida durante o período de tratamento com capecitabina.
Fertilidade
Não existem dados sobre o Xeloda e o seu impacto na fertilidade. Os estudos principais do Xeloda incluíram mulheres com potencial para engravidar e homens apenas se estivessem dispostos a utilizar métodos contracetivos adequados para evitar a gravidez durante o estudo e durante um período razoável a partir de então.
Foram observados efeitos na fertilidade em estudos com animais (ver secção 5.3).
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
A capecitabina tem uma influência ligeira a moderada na capacidade de conduzir ou utilizar máquinas. Capecitabina pode induzir tonturas, fadiga e náuseas.
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
O perfil de segurança geral da capecitabina é baseado em dados de mais de 3.000 pacientes tratados com capecitabina isoladamente ou com capecitabina em combinação com diferentes regimes de quimioterapia em múltiplas indicações. Os perfis de segurança da monoterapia com capecitabina em populações de pacientes com câncer de mama metastático, câncer colorretal metastático e câncer de cólon adjuvante são semelhantes. Consulte a seção 5.1 para obter detalhes sobre os estudos principais, incluindo desenhos de estudos e os principais resultados de eficácia.
As reações adversas medicamentosas (RAMs) relacionadas ao tratamento mais comumente relatadas e / ou clinicamente relevantes foram distúrbios gastrointestinais (especialmente diarreia, náuseas, vômitos, dor abdominal, estomatite), síndrome mão-pé (eritrodisestesia palmo-plantar), fadiga, astenia, anorexia, cardiotoxicidade, piora da função renal em que a função já estava previamente prejudicada e trombose / embolia.
Resumo das reações adversas em forma tabular
As RAMs consideradas pelo investigador como possível, provável ou remotamente relacionadas à administração de capecitabina estão listadas na Tabela 4 para tomar capecitabina sozinha e na Tabela 5 para tomar capecitabina em combinação com diferentes regimes de quimioterapia em indicações múltiplas. Os seguintes termos são usados para classificar ADRs de acordo com sua frequência: muito comuns (≥ 1/10), comuns (≥ 1/100,
Monoterapia com capecitabina:
A Tabela 4 lista as RAMs associadas ao uso de monoterapia com capecitabina com base em uma análise agrupada de dados de segurança de três estudos principais, incluindo mais de 1900 pacientes (estudos M66001, SO14695 e SO14796). As RAMs foram incluídas no grupo de frequência específico de acordo com a "incidência geral decorrente da análise agregada".
Tabela 4 Resumo das RAMs relacionadas relatadas em pacientes tratados com monoterapia com capecitabina.
Capecitabina em terapia combinada:
A Tabela 5 lista as RAMs relacionadas ao uso de capecitabina em combinação com diferentes regimes de quimioterapia em várias indicações com base em dados de segurança de mais de 3.000 pacientes. As RAMs foram incluídas no grupo de frequência específico (muito comum ou comum) com base na incidência mais alta observados nos estudos clínicos principais e apenas se forem adicionais aos observados com a monoterapia com capecitabina ou se pertencer a um grupo de frequência mais elevada do que a monoterapia com capecitabina (ver tabela 4). As RAMs pouco frequentes notificadas para a capecitabina em terapêutica combinada são consistentes com as RAMs notificadas para a capecitabina em monoterapia ou monoterapia com medicamentos combinados (na literatura e / ou no respetivo resumo das características do medicamento).
Algumas das RAMs são reações frequentemente observadas com o medicamento combinado (por exemplo, neuropatia sensorial periférica com docetaxel ou oxaliplatina, hipertensão com bevacizumab); no entanto, o agravamento induzido pela terapia com capecitabina não pode ser excluído.
Tabela 5 Resumo das RAMs relatadas em pacientes tratados com capecitabina em terapia de combinação, além daquelas observadas com capecitabina sozinha ou observadas em um grupo de frequência mais alta do que a capecitabina sozinha.
+ Para cada período, a frequência foi calculada com base nas ADRs de todas as séries. Para os termos marcados com "+", a frequência foi calculada com base em ADRs de grau 3-4. As RAMs foram incluídas com base na incidência mais alta observada nos ensaios clínicos de terapia combinada principal.
Descrição de uma seleção de reações adversas
Síndrome mão-pé (ver seção 4.4):
Em estudos de monoterapia com capecitabina (incluindo estudos de terapia adjuvante em câncer de cólon, tratamento de câncer colorretal metastático e tratamento de câncer de mama), com 1250 mg / m2 de capecitabina duas vezes ao dia nos dias 1 a 14 a cada três semanas, síndrome mão-pé de qualquer grau foi observada com frequência variando de 53% a 60%; no braço capecitabina / docetaxel para o tratamento do câncer de mama metastático, a frequência foi de 63%. Em terapia combinada com capecitabina, com capecitabina 1000 mg / m2 duas vezes ao dia nos dias 1 a 14 a cada três semanas, qualquer grau de síndrome mão-pé foi observado com uma frequência variando de 22% a 30%.
Como parte de uma meta-análise em 14 ensaios clínicos, com dados de mais de 4.700 pacientes tratados com capecitabina em monoterapia ou capecitabina em combinação com diferentes regimes de quimioterapia em múltiplas indicações (câncer de cólon, colorretal, gástrico e de mama), síndrome mão-pé de qualquer grau ocorreu em 2.066 pacientes (43%) após uma mediana de 239 dias (IC 95%: 201, 288) desde o início do tratamento com capecitabina. Em todos os estudos combinados, houve uma "associação estatisticamente significativa entre as seguintes covariáveis e um risco aumentado de desenvolver síndrome mão-pé: aumento na dose inicial de capecitabina (grama), diminuição na dose cumulativa de capecitabina (0,1 * kg), aumento relativo intensidade da dose nas primeiras 6 semanas, duração aumentada do tratamento do estudo (semanas), idade avançada (incrementos de 10 anos), sexo feminino e bom estado de desempenho ECOG inicial (0 vs ≥1).
Diarreia (ver seção 4.4):
A capecitabina pode induzir o aparecimento de diarreia, que foi observada em até 50% dos pacientes.
Os resultados de uma meta-análise de 14 estudos clínicos com dados de mais de 4.700 pacientes tratados com capecitabina mostraram que em todos os estudos combinados houve uma "associação estatisticamente significativa entre as seguintes covariáveis e um risco aumentado de desenvolver diarreia: dose inicial aumentada de capecitabina (grama), aumento da duração do tratamento do estudo (semanas), idade avançada (incrementos de 10 anos) e sexo feminino. Uma associação estatisticamente significativa foi observada entre as seguintes covariáveis e uma redução no risco de desenvolver diarreia: aumento na dose cumulativa de capecitabina (0,1 * kg) e aumento na intensidade da dose relativa nas primeiras 6 semanas.
Cardiotoxicidade (ver seção 4.4):
Além dos ADRs descritos nas Tabelas 4 e 5, com base em uma "análise agrupada de dados de segurança clínica de 7 estudos clínicos, incluindo 949 pacientes (2 estudos de fase III e 5 de fase II em câncer colorretal metastático) e em câncer de mama metastático), as seguintes RAMs com incidência inferior a 0,1% foram observadas em associação com o uso de capecitabina isoladamente: cardiomiopatia, insuficiência cardíaca, morte súbita e extrassístoles ventriculares.
Encefalopatia:
Além das RAMs descritas nas tabelas 4 e 5, com base na análise combinada acima mencionada de dados de segurança clínica de 7 estudos clínicos, o uso de capecitabina isoladamente também foi associado a encefalopatia, com uma incidência de menos de 0,1%.
Populações especiais
Pacientes mais velhos (ver seção 4.2):
Uma "análise de dados de segurança em pacientes ≥ 60 anos de idade tratados com capecitabina em monoterapia e uma" análise de pacientes tratados com a combinação terapêutica de capecitabina e docetaxel mostraram um aumento da incidência de reações adversas de grau 3 e 4 relacionadas e relacionadas ao tratamento reações adversas graves em comparação com pacientes com menos de 60 anos de idade. Além disso, os doentes com ≥60 anos de idade tratados com capecitabina e docetaxel interromperam o tratamento prematuramente devido a reações adversas mais frequentes do que os doentes com menos de 60 anos.
Os resultados de uma meta-análise de 14 estudos clínicos com dados de mais de 4.700 pacientes tratados com capecitabina mostraram que em todos os estudos combinados houve uma "associação estatisticamente significativa entre" o avanço da idade (incrementos de 10 anos) e um risco aumentado de desenvolver síndrome mão-pé e diarreia, e um risco reduzido de desenvolver neutropenia.
Sexo
Os resultados de uma meta-análise de 14 ensaios clínicos com dados de mais de 4.700 pacientes tratados com capecitabina mostraram que em todos os estudos combinados houve uma "associação estatisticamente significativa entre o sexo feminino e um risco aumentado de desenvolver a síndrome. Mão-pé e diarreia e um risco reduzido de desenvolver neutropenia.
Doentes com compromisso renal (ver secções 4.2, 4.4 e 5.2):
Uma análise dos dados de segurança em pacientes tratados com monoterapia com capecitabina (câncer colorretal) com insuficiência renal no início do estudo mostrou um aumento na incidência de reações adversas relacionadas ao tratamento de grau 3 e 4 em comparação com pacientes com insuficiência renal normal (36% em pacientes sem insuficiência renal n = 268 vs 41% no comprometimento leve n = 257 e 54% no moderado n = 59, respectivamente) (ver seção 5.2). Um aumento na taxa de redução da dose (44%) foi observado em pacientes com função renal moderadamente comprometida vs 33% e 32% em pacientes com insuficiência renal leve ou sem comprometimento renal e um aumento na descontinuação prematura do tratamento (21% de interrupções durante os dois primeiros ciclos) vs 5% e 8% em pacientes com pouca ou nenhuma insuficiência renal.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas ocorridas após a autorização do medicamento é importante porque permite a monitorização contínua da relação benefício / risco do medicamento. Os profissionais de saúde são solicitados a notificar quaisquer suspeitas de reações adversas através do sistema nacional de notificação www .agenziafarmaco.gov .it / it / responsável.
04.9 Overdose
As manifestações de sobredosagem aguda incluem náuseas, vómitos, diarreia, mucosite, irritação gastrointestinal e hemorragia, bem como depressão da medula óssea. O manejo clínico da sobredosagem deve ser feito por meio de terapia convencional e intervenção médica de suporte, a fim de corrigir as manifestações clínicas presentes e prevenir quaisquer possíveis complicações decorrentes.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: citostático (antimetabólito).
Código ATC: L01BC06.
A capecitabina é um carbamato de fluoropirimidina não citotóxico que atua como um precursor administrável por via oral da forma citotóxica 5-fluorouracil (5-FU). A capecitabina é ativada por meio de várias etapas enzimáticas (ver seção 5.2). A enzima envolvida na conversão final em 5-FU, timidina fosforilase (ThyPase), é encontrada em tecidos tumorais, mas também em tecidos normais, embora geralmente em uma concentração mais baixa. Em modelos de tumor de xenoenxerto humano, a capecitabina demonstrou ter um efeito sinérgico em combinação com docetaxel, que pode estar relacionado com a hiperregulação da timidina fosforilase pelo docetaxel.
Foi observado que o metabolismo do 5-FU na via anabólica bloqueia a reação de metilação do ácido desoxiuridil em ácido timidil, interferindo assim na síntese do ácido desoxirribonucléico (DNA). A incorporação de 5-FU também leva à inibição do RNA e da síntese de proteínas. Uma vez que o DNA e o RNA são essenciais para a divisão e crescimento celular, o 5-FU pode resultar em deficiência de timidina, que causa crescimento desequilibrado e morte celular. Os efeitos da privação de DNA e RNA são particularmente marcados em células que crescem mais rápido e metabolizam o 5-FU mais rápido.
Câncer de cólon e colorretal:
Monoterapia com capecitabina no tratamento adjuvante do câncer de cólon
Dados de um ensaio clínico multicêntrico, randomizado e controlado de fase III em pacientes com câncer de cólon em estágio III (Dukes C) apóiam o uso de capecitabina para terapia adjuvante em pacientes com câncer de cólon (X-ACT Study, M66001). Neste estudo, 1987 os pacientes foram randomizados para tratamento com capecitabina (1250 mg / m2 duas vezes ao dia por 2 semanas, seguido por 1 semana de folga, como ciclos de 3 semanas por 24 semanas) ou 5-FU e leucovorina (esquema da Mayo Clinic: 20 mg / m2 leucovorina IV seguido por bolus de 5-FU IV de 425 mg / m2, nos dias 1 a 5, a cada 28 dias por 24 semanas). Capecitabina foi pelo menos equivalente a 5-FU / LV IV na sobrevida livre de doença na população por protocolo (HR 0,92; IC 95%: 0,80-1,06). A sobrevida livre de doença e a sobrevida global mostraram um HR de 0,88 (IC 95%: 0,77-1,01; p = 0,068) e 0,86 (IC 95%: 0,74-1,01; p = 0,060), respectivamente. O acompanhamento médio no momento da análise foi de 6,9 anos. Em uma análise multivariada de Cox previamente planejada, foi demonstrada a superioridade da capecitabina em relação ao bolus de 5-FU / VE. Os seguintes fatores foram predefinidos na análise estatística para inclusão no modelo: idade, tempo desde a cirurgia até a randomização, sexo, níveis basais de CEA, linfonodos basais e país. Em toda a população randomizada, a capecitabina mostrou ser superior ao 5-FU / LV tanto em termos de sobrevida livre de doença (HR: 0,849; IC 95%: 0,739-0,976; p = 0,0212) e em termos de sobrevida geral (HR : 0,828; IC 95%: 0,705-0,971; p = 0,0203).
Terapia de combinação no tratamento adjuvante do câncer de cólon
Dados de um ensaio clínico multicêntrico, randomizado e controlado de fase III em pacientes com câncer de cólon em estágio III (Dukes C) apóiam o uso de capecitabina em combinação com oxaliplatina (XELOX) para tratamento adjuvante em pacientes com câncer de cólon (Estudo NO16968).Neste estudo, 944 pacientes foram randomizados para tratamento com capecitabina (1000 mg / m2 duas vezes ao dia por 2 semanas, seguido por 1 semana de folga, como cursos de 3 semanas por 24 semanas) em combinação com oxaliplatina (130 mg / m2 por infusão intravenosa por 2 horas no dia 1 a cada 3 semanas); 942 pacientes foram randomizados para bolus de 5-FU e leucovorina. Na análise primária para DFS na população ITT, XELOX mostrou ser significativamente superior a 5-FU / LV (HR = 0,80, IC de 95% = [0,69; 0,93]; p = 0,0045). A taxa de DFS foi de 71% no braço XELOX em comparação com 67% no braço 5-FU / LV. A análise realizada para o desfecho secundário de RFS suporta esses resultados com um HR de 0,78 (IC 95% = [0,67; 0,92]; p = 0,0024) no braço XELOX em comparação com o braço 5-FU / LV. XELOX demonstrou uma tendência de superioridade em termos de OS com um HR de 0,87 (IC 95% = [0,72; 1,05]; p = 0,1486), o que se traduz em uma redução de 13% no risco de morte. OS de 5 anos foi de 78% para XELOX versus 74% para 5-FU / LV. Os dados de eficácia são baseados em um tempo médio de observação de 59 meses para OS e 57 meses para DFS. A taxa de retirada do estudo para eventos adversos foi maior no braço XELOX (21%) do que no braço de monoterapia 5-FU / LV (9%) na população ITT.
Monoterapia com capecitabina em câncer colorretal metastático
Dados de dois ensaios clínicos de fase III controlados, multicêntricos, randomizados e controlados de forma semelhante (SO14695: SO14796) apóiam o uso de capecitabina para o tratamento de primeira linha do câncer colorretal metastático. Nestes estudos, 603 pacientes foram randomizados para tratamento com capecitabina (1250 mg / m2 duas vezes ao dia por 2 semanas, seguido por um descanso de 1 semana, e administrado em ciclos de 3 semanas). 604 pacientes foram randomizados para tratamento com 5-FU e leucovorina (regime Mayo: 20 mg / m2 iv leucovorina seguido por 425 mg / m2 em bolus intravenoso de 5-FU, nos dias 1 a 5, a cada 28 dias). investigador) foram: 25,7% (capecitabina) versus 16,7% (regime Mayo); p
Terapia combinada no tratamento de primeira linha do câncer colorretal metastático
Os dados de um ensaio clínico multicêntrico, randomizado e controlado de fase III (NO16966) apóiam o uso de capecitabina em combinação com oxaliplatina ou em combinação com oxaliplatina e bevacizumabe para o tratamento de primeira linha de câncer colorretal metastático. O estudo incluiu duas partes: as duas iniciais - parte do braço em que 634 pacientes foram randomizados para dois regimes de tratamento diferentes, ou seja, XELOX ou FOLFOX-4, e uma parte fatorial 2x2 subsequente em que 1401 pacientes foram randomizados para quatro regimes de tratamento diferentes. tratamento, ou seja, XELOX mais placebo, FOLFOX-4 mais placebo, XELOX mais bevacizumabe e FOLFOX-4 mais bevacizumabe Consulte a tabela 6 para os regimes de tratamento.
Tabela 6 Regimes de tratamento no estudo NO16966 (mCRC)
Na comparação geral, a não inferioridade dos braços contendo XELOX em comparação com aqueles contendo FOLFOX-4 foi demonstrada em termos de sobrevida livre de progressão na população de pacientes elegíveis e na população com intenção de tratar (ver tabela 7). Os resultados indicam que XELOX é equivalente a FOLFOX-4 em termos de sobrevida global (ver Tabela 7). A comparação de XELOX mais bevacizumabe versus FOLFOX-4 mais bevacizumabe consistiu em uma "análise exploratória pré-planejada. Ao comparar esses subgrupos de tratamento, XELOX mais bevacizumabe foi semelhante a FOLFOX-4 mais bevacizumabe em termos de sobrevida livre de progressão (taxa de risco 1,01; IC 97,5%: 0,84 - 1,22). O acompanhamento médio no momento das análises primárias na população com intenção de tratar foi de 1,5 anos; os dados derivados de realizados após um ano adicional de acompanhamento também estão incluídos na tabela 7 . A análise de PFS durante o tratamento não confirmou, no entanto, os resultados da análise de PFS geral e OS: a razão de risco de XELOX versus FOLFOX -4 foi de 1,24 com IC de 97,5%: 1,07-1,44. Embora as análises de sensibilidade mostrem essas diferenças no planejamento do regime e no tempo de avaliação do tumor afetam a análise de PFS em curso do tratamento, nenhuma explicação definitiva para isso foi encontrada resultado.
Tabela 7 Principais resultados de eficácia para a análise de não inferioridade do Estudo NO16966
* PPE = população de pacientes elegíveis; ** ITT = população com intenção de tratar.
Em um estudo de fase III, randomizado e controlado (CAIRO), o efeito do uso de capecitabina em uma dose inicial de 1000 mg / m2 por 2 semanas a cada 3 semanas em combinação com irinotecano para o tratamento de primeira linha foi investigado. De pacientes com metástase colorretal Câncer. 820 pacientes foram randomizados para receber tratamento sequencial (n = 410) ou de combinação (n = 410). O tratamento sequencial consistiu em um tratamento de primeira linha com capecitabina (1250 mg / m2 duas vezes ao dia por 14 dias), uma segunda linha com irinotecano (350 mg / m2 no dia 1) e uma terceira linha com a combinação de capecitabina. (1000 mg / m2 duas vezes ao dia por 14 dias) e oxaliplatina (130 mg / m2 no dia 1). O tratamento combinado consistiu em tratamento de primeira linha com capecitabina (1000 mg / m2 duas vezes ao dia por 14 dias) combinado com irinotecano (250 mg / m2 no dia 1 ) (XELIRI) e uma segunda linha com capecitabina (1000 mg / m2 duas vezes ao dia por 14 dias) mais oxaliplatina (130 mg / m2 no dia 1). Foram administradas em intervalos de 3 semanas. No tratamento de primeira linha, a progressão média - a sobrevida livre na população com intenção de tratar foi de 5,8 meses (IC 95%; 5,1 - 6,2 meses) para monoterapia com capecitabina e 7,8 meses (IC 95%: 7,0 - 8,3 meses; p = 0,0002) para XELIRI. isto foi associado a um aumento da incidência de toxicidade gastrointestinal e neutropenia durante o tratamento de primeira linha com XELIRI (26% e 11% para XELIRI e capecitabina de primeira linha, respectivamente).
Em três estudos randomizados em pacientes com câncer colorretal metastático, o regime XELIRI foi comparado com 5-FU + irinotecano (FOLFIRI). Os regimes XELIRI incluíram capecitabina 1000 mg / m2 duas vezes ao dia nos dias 1 a 14 de um ciclo de três semanas combinado com irinotecano 250 mg / m2 no dia 1. No estudo maior (BICC-C), os pacientes foram randomizados para rótulo aberto tratamento com FOLFIRI (n = 144), bolus 5-FU (mIFL) (n = 145) ou XELIRI (n = 141) e posteriormente randomizado para celecoxibe duplo-cego ou placebo. A mediana de PFS foi de 7,6 meses para FOLFIRI, 5,9 meses para mIFL (p = 0,004 para comparação com FOLFIRI) e 5,8 meses para XELIRI (p = 0,015). A SG média foi de 23,1 meses para FOLFIRI, 17,6 meses para mIFL (p = 0,09) e 18,9 meses para XELIRI (p = 0,27). Os pacientes tratados com XELIRI experimentaram toxicidade gastronintestinal excessiva em comparação com aqueles tratados com FOLFIRI (48% e 14% de diarreia para XELIRI e FOLFIRI, respectivamente).
No estudo EORTC, os pacientes foram randomizados para tratamento aberto com FOLFIRI (n = 41) ou XELIRI (n = 44) e posteriormente randomizados para celecoxibe duplo-cego ou placebo. PFS mediana e sobrevida geral (OS) foram menores para XELIRI em comparação com FOLFIRI (PFS 5,9 versus 9,6 meses e OS 14,8 versus 19,9 meses); além disso, taxas excessivas de diarreia foram relatadas em pacientes recebendo o regime XELIRI (41% XELIRI; 5,1% FOLFIRI).
No estudo publicado pela Skof et al., os pacientes foram randomizados para receber FOLFIRI ou XELIRI. A taxa de resposta geral foi de 49% no braço XELIRI e 48% no braço FOLFIRI (p = 0,76). No final do tratamento, 37% dos pacientes no braço XELIRI e 26% dos pacientes no braço FOLFIRI não tinham evidência de doença (p = 0,56). A toxicidade foi semelhante entre os tratamentos, com exceção da neutropenia, que foi mais comumente relatada em pacientes tratados com FOLFIRI.
Montagnani et al. eles usaram os resultados dos três estudos acima mencionados para fornecer uma "análise global dos estudos randomizados comparando os regimes terapêuticos FOLFIRI e XELIRI no tratamento de mCRC." Uma redução significativa no risco de progressão da doença foi associada ao tratamento com FOLFIRI (HR 0,76; IC 95%: 0,62-0,95; p
Dados de um ensaio clínico randomizado (Souglakos et al., 2012) comparações entre FOLFIRI + bevacizumab e XELIRI + bevacizumab não mostraram diferenças significativas em termos de PFS e OS entre os tratamentos. Os pacientes foram randomizados para tratamento com FOLFIRI mais bevacizumabe (Braço A, n = 167) ou XELIRI mais bevacizumabe (Braço B, n = 166). Para o Braço B, o regime XELIRI usou capecitabina 1000 mg / m2 duas vezes ao dia por 14 dias + irinotecano 250 mg / m2 no dia 1. Para tratamento com FOLFIRI-Bev e tratamento com XELIRI-Bev, respectivamente, a sobrevida livre de progressão mediana ( PFS), sobrevida global e taxas de resposta foram as seguintes: 10,0 meses e 8,9 meses (p = 0,64); 25,7 meses e 27,5 meses (p = 0,55); 45,5% e 39,8% (p = 0,32). Os pacientes tratados com XELIRI + bevacizumabe relataram uma incidência significativamente maior de diarreia, neutropenia febril e reações cutâneas mão-pé em comparação com pacientes tratados com FOLFIRI + bevacizumabe com atrasos de tratamento significativamente aumentados, reduções de dose e interrupções de tratamento.
Dados de um estudo de fase II, multicêntrico, randomizado e controlado (AIO KRK 0604) apóiam o uso de capecitabina em uma dose inicial de 800 mg / m2 por 2 semanas a cada 3 semanas em combinação com irinotecano e bevacizumabe para tratamento. pacientes com câncer colorretal metastático.
120 pacientes foram randomizados para um regime XELIRI modificado com capecitabina 800 mg / m2 duas vezes ao dia por duas semanas, seguido por 7 dias de repouso), irinotecano (200 mg / m2 como uma infusão de 30 minutos no dia 1 a cada 3 semanas) e bevacizumabe (7,5 mg / kg infundidos por 30 a 90 minutos no dia 1 a cada 3 semanas); 127 pacientes foram randomizados para tratamento com capecitabina (1000 mg / m2 duas vezes ao dia por duas semanas seguidas por 7 dias de repouso), oxaliplatina (130 mg / m2 como uma infusão de 2 horas no dia 1 a cada 3 semanas) e bevacizumabe (7,5 mg / kg infundido por 30 a 90 minutos no dia 1 a cada 3 semanas). Após uma duração média de acompanhamento para a população do estudo de 26,2 meses, as respostas ao tratamento foram as seguintes:
Tabela 8 Resultados de eficácia para o estudo AIO KRK
Terapia de combinação no tratamento de segunda linha do câncer colorretal metastático
Dados de um ensaio clínico de Fase III, multicêntrico, randomizado e controlado (NO16967) apóiam o uso de capecitabina em combinação com oxaliplatina para o tratamento de segunda linha de câncer colorretal metastático. Neste estudo, 627 pacientes com câncer colorretal metastático que receberam tratamento anterior com irinotecano em combinação com um regime à base de fluoropirimidina como tratamento de primeira linha foram randomizados para tratamento com XELOX ou FOLFOX-4. Para o regime de dosagem de XELOX e FOLFOX-4 (sem a adição de placebo ou bevacizumabe), consulte a tabela 6. XELOX demonstrou ser não inferior ao FOLFOX-4 em termos de sobrevida livre de progressão no protocolo e na população com intenção de tratar (ver tabela 9). Os resultados indicam que XELOX é equivalente a FOLFOX -4 em termos de sobrevida global (ver tabela 9). O acompanhamento médio no momento das análises primárias na população com intenção de tratar era em 2,1 anos; os dados das análises realizadas após mais 6 meses de acompanhamento também estão incluídos na tabela 9.
Tabela 9 Principais resultados de eficácia para a análise de não inferioridade do estudo NO16967
* PPP = população por protocolo; ** ITT = população com intenção de tratar.
Câncer gástrico avançado:
Dados de um ensaio clínico multicêntrico, randomizado e controlado de fase III em pacientes com câncer gástrico avançado apóiam o uso de capecitabina no tratamento de primeira linha do câncer gástrico avançado (ML17032). Neste estudo, 160 pacientes foram randomizados. Tratamento com capecitabina ( 1000 mg / m2 duas vezes ao dia por 2 semanas seguidas por 7 dias de repouso) e cisplatina (80 mg / m2 como uma infusão de 2 horas a cada 3 semanas). Um total de 156 pacientes foram randomizados para tratamento com 5-FU (800 mg / m2 por dia, como uma infusão contínua do dia 1 ao dia 5 a cada 3 semanas) e cisplatina (80 mg / m2 como uma infusão de 2 horas no dia 1 a cada 3 semanas). Capecitabina em combinação com cisplatina demonstrou não inferioridade a 5-FU em combinação com cisplatina em termos de sobrevida livre de progressão na análise por protocolo (HR 0,81; IC 95%: 0,63-1,04). A sobrevida livre de progressão mediana foi de 5,6 meses (capecitabina + cisplatina) em comparação com 5,0 meses (5-FU + cisplatina). A razão de risco para a duração da sobrevida (sobrevida global) foi semelhante à razão de risco para a sobrevida livre de progressão (HR 0,85; IC 95%: 0,64-1,13). A duração média de sobrevivência foi de 10,5 meses (capecitabina + cisplatina) em comparação com 9,3 meses (5-FU + cisplatina).
Dados de um ensaio clínico de fase III, multicêntrico, randomizado, comparando capecitabina a 5-FU e oxaliplatina e cisplatina em pacientes com câncer gástrico avançado, apóiam o uso de capecitabina no tratamento de primeira linha de câncer gástrico avançado (REAL-2). estudo, 1.002 pacientes foram randomizados com um design fatorial 2x2 para um dos seguintes 4 braços:
- ECF: epirrubicina (50 mg / m2 em bolus no dia 1 a cada 3 semanas), cisplatina (60 mg / m2 em infusão de 2 horas no dia 1 a cada 3 semanas) e 5-FU (200 mg / m2 administrado diariamente à medida que a infusão continua através do cateter central).
- ECX: epirrubicina (50 mg / m2 em bolus no dia 1 a cada 3 semanas), cisplatina (60 mg / m2 em infusão de 2 horas no dia 1 a cada 3 semanas) e capecitabina (625 mg / m2 duas vezes ao dia como tratamento contínuo).
- EOF: epirrubicina (50 mg / m2 em bolus no dia 1 a cada 3 semanas), oxaliplatina (130 mg / m2 em infusão de 2 horas no dia 1 a cada 3 semanas) e 5-FU (200 mg / m2 administrado diariamente à medida que a infusão continua através do cateter central).
- EOX: epirrubicina (50 mg / m2 em bolus no dia 1 a cada 3 semanas), oxaliplatina (130 mg / m2 em infusão de 2 horas no dia 1 a cada 3 semanas) e capecitabina (625 mg / m2 duas vezes ao dia como tratamento contínuo).
As análises de eficácia primária na população por protocolo demonstraram não inferioridade na sobrevida geral para regimes contendo capecitabina em comparação com regimes baseados em 5-FU (HR 0,86; IC 95%: 0,8-0,0, 99) e para regimes contendo oxaliplatina comparados a regimes baseados em cisplatina (HR 0,92; IC 95%: 0,80-1,1). A sobrevida global mediana foi de 10,9 meses nos regimes baseados em capecitabina e 9,6 meses naqueles contendo 5-FU. A sobrevida global mediana foi de 10,0 meses nos regimes baseados em cisplatina e 10,4 meses nos regimes baseados em oxaliplatina.
Capecitabina também tem sido usada em combinação com oxaliplatina no tratamento do câncer gástrico avançado. Estudos com monoterapia com capecitabina indicam que a capecitabina exibe atividade no câncer gástrico avançado.
Câncer avançado de estômago, cólon e colorretal: meta-análise
Uma meta-análise de seis estudos clínicos (estudos SO14695, SO14796, M66001, NO16966, NO16967, M17032) apóia o uso de capecitabina como substituto do 5-FU sozinho e no tratamento combinado de câncer gastrointestinal. A análise agrupada inclui 3097 pacientes tratados com regimes contendo capecitabina e 3.074 pacientes tratados com regimes contendo 5-FU. A sobrevida global média foi de 703 dias (IC 95%: 671; 745) em pacientes tratados com regimes contendo capecitabina e 683 dias (IC 95%: 646; 715) naqueles tratados com regimes contendo 5-FU. A razão de risco para a sobrevida global foi de 0,94 (IC 95%: 0,89; 1,00, p = 0,0489), indicando que os regimes contendo capecitabina não são inferiores aos que contêm 5-FU.
Câncer de mama
Terapia de combinação com capecitabina e docetaxel no câncer de mama localmente avançado ou metastático
Dados de um ensaio clínico de fase III, multicêntrico, randomizado e controlado apóiam o uso de capecitabina em combinação com docetaxel para o tratamento de pacientes com câncer de mama localmente avançado ou metastático após falha da quimioterapia citotóxica que incluiu uma "antraciclina. Neste estudo, 255 pacientes foram randomizados para tratamento com capecitabina (1250 mg / m2 duas vezes ao dia por 2 semanas, seguido por um período de descanso de 1 semana e docetaxel 75 mg / m2 como infusão intravenosa de 1 hora a cada 3 semanas). 256 pacientes foram randomizados para tratamento com docetaxel sozinho (100 mg / m2 como uma infusão intravenosa de 1 hora a cada 3 semanas). A sobrevivência foi superior no braço da combinação capecitabina + docetaxel (p = 0,0126). A sobrevida média foi de 442 dias (capecitabina + docetaxel) em comparação com 352 dias (docetaxel sozinho). As taxas de resposta objetiva geral em toda a população randomizada (avaliação do investigador) foram: 41,6% (capecitabina + docetaxel) vs. 29,7% (docetaxel sozinho); p = 0,0058. O tempo de progressão da doença foi superior no braço da combinação capecitabina + docetaxel ( p
Monoterapia com capecitabina após falha do taxano e de uma quimioterapia contendo antraciclina e quando a terapia com antraciclina não é indicada
Dados de dois ensaios clínicos multicêntricos de Fase II suportam o uso de monoterapia com capecitabina para o tratamento de pacientes que estão progredindo após falha da quimioterapia que incluiu taxanos e uma antraciclina, ou para os quais nenhuma terapia adicional é indicada. Antraciclinas. Nestes estudos, um total de 236 pacientes foram tratados com capecitabina (1250 mg / m2 duas vezes ao dia por 2 semanas seguidas por um período de descanso de 1 semana). As taxas de resposta objetiva geral (avaliação do investigador) foram de 20% (primeiro estudo) e 25% (segundo estudo) Tempo médio para a progressão foi de 93 e 98 dias. A sobrevida média foi de 384 e 373 dias.
Todas as indicações:
Uma meta-análise de 14 ensaios clínicos com dados de mais de 4.700 pacientes tratados com capecitabina isoladamente ou em combinação com diferentes regimes de quimioterapia em várias indicações (câncer de cólon, colorretal, gástrico e de mama) mostrou sobrevida geral mais prolongada em pacientes tratados com capecitabina que desenvolveram síndrome mão-pé do que em pacientes que não desenvolveram: sobrevida global mediana 1100 dias (IC 95%: 1007, 1200) vs 691 dias (IC 95%: 638; 754) com uma razão de risco de 0,61 (IC 95%: 0,56 , 0,66).
População pediátrica:
A Agência Europeia de Medicamentos dispensou a obrigação de realizar estudos com Xeloda em todas as subclasses da população pediátrica no adenocarcinoma do cólon e retal, adenocarcinoma gástrico e cancro da mama (ver secção 4.2 para informação sobre “utilização pediátrica).
05.2 Propriedades farmacocinéticas
A farmacocinética da capecitabina foi avaliada ao longo de um intervalo de doses de 502-3514 mg / m2 / dia. Os parâmetros de capecitabina, 5 "-desoxi-5-fluorocitidina (5" -DFCR) e 5 "-desoxi-5-fluorouridina (5" DFUR) medidos nos dias 1 e 14 foram semelhantes. A AUC de 5-FU no dia 14 foi 30-35% maior. A redução da dose de capecitabina diminui a exposição sistêmica ao 5-FU de uma forma mais proporcional à dose devido à farmacocinética não linear do metabólito ativo.
Absorção
Após a administração oral, a capecitabina é completa e rapidamente absorvida; subsequentemente, é completamente convertido nos metabólitos 5 "-DFCR e 5" -DFUR. A administração com alimentos diminui a taxa de absorção da capecitabina, mas causa apenas um efeito menor na AUC de 5 "-DFUR e na AUC do metabólito subsequente 5-FU. A uma dose de 1250 mg / m2 no dia 14 administrada após as refeições, as concentrações plasmáticas máximas (Cmax em mcg / ml) de capecitabina, 5 "-DFCR, 5" -DFUR, 5-FU e FBAL foram respectivamente 4,67 - 3,05 - 12,1 - 0,95 e 5,46. O tempo para atingir as concentrações plasmáticas máximas (Tmax em horas) foi 1,50 - 2,00 - 2,00 - 2,00 e 3,34. Os valores de AUC0- ∞ em mcg • h / ml foram 7,75 - 7,24 - 24,6 - 2,03 e 36,3.
Distribuição
Estudos de plasma humano realizados em vitro mostraram que capecitabina, 5 "DFCR, 5" -DFUR e 5-FU estão ligados a proteínas, principalmente albumina, em porcentagens de 54%, 10%, 62% e 10% respectivamente.
Biotransformação
A capecitabina é primeiro metabolizada pela carboxilesterase hepática em 5 "-DFCR, que é subsequentemente convertido em 5" -DFUR pela citidina desaminase, principalmente localizada no fígado e nos tecidos tumorais. Ocorre então uma "ativação catalítica adicional de 5" -DFUR pela timidina fosforilase (ThyPase). As enzimas envolvidas na ativação catalítica estão presentes em tecidos tumorais, mas também em tecidos saudáveis, embora geralmente em quantidades menores.A biotransformação enzimática sequencial da capecitabina em 5-FU leva a maiores concentrações nos tecidos neoplásicos. Em cânceres colorretais, a geração de 5-FU parece estar amplamente localizada nas células do estroma tumoral. Após a administração oral de capecitabina a pacientes com câncer colorretal, a proporção da concentração de 5-FU em cânceres colorretais para tecidos adjacentes foi de 3,2 (variando de 0,9 a 8,0). A proporção da concentração de 5-FU no tumor para o plasma foi de 21,4 (variando de 3,9 a 59,9, n = 8), enquanto a proporção no tecido saudável para o plasma foi de 8,9 (com variação de 3,0 a 25,8, n = 8). A atividade da timidina fosforilase foi medida e encontrada ser 4 vezes maior no câncer colorretal primário do que os valores relatados no tecido normal adjacente. Com base em estudos de imunohistoquímica, a timidina fosforilase parece estar amplamente localizada nas células do estroma tumoral.
5-FU é subsequentemente catabolizado pela enzima dihidropirimidina desidrogenase (DPD) em dihidro-5-fluorouracil (FUH2), que é muito menos tóxico. A dihidropirimidase atua no anel de pirimidina para obter ácido 5-fluoro-ureidopropiônico (FUPA). Finalmente, b -ureido-propionase transforma FUPA em a-fluoro-b-alanina (FBAL) que é eliminada na urina. A atividade da dihidropirimidina desidrogenase (DPD) é o fator limitante crítico. A deficiência de DPD pode induzir “aumento da toxicidade da capecitabina (ver secções 4.3 e 4.4).
Eliminação
A meia-vida de eliminação (t½ em horas) da capecitabina, 5 "-DFCR, 5" -DFUR, 5-FU e FBAL foi de 0,85 - 1,11 - 0,66 - 0,76 e 3, respectivamente, 23. A capecitabina e seus metabólitos são eliminados principalmente na urina; 95,5% da dose administrada de capecitabina foi recuperada na urina.A excreção fecal é mínima (2,6%). O principal metabólito excretado na urina é o FBLA, que representa 57% da dose administrada. Aproximadamente 3% da dose administrada é excretada na urina como fármaco inalterado.
Terapia de associação
Estudos de fase I que avaliaram os efeitos da capecitabina na farmacocinética do docetaxel ou paclitaxel e vice-versa mostraram que não há efeito da capecitabina na farmacocinética do docetaxel ou paclitaxel (Cmax e AUC) e que não há efeito do docetaxel ou paclitaxel no a farmacocinética de 5 "-DFUR.
Farmacocinética em populações particulares de pacientes
Uma análise farmacocinética populacional foi realizada após o tratamento com capecitabina administrada em uma dose de 1250 mg / m2 duas vezes ao dia em 505 pacientes com câncer colorretal. Sexo, presença ou ausência de metástases hepáticas na linha de base, status de desempenho de Karnofsky, bilirrubina total, albumina sérica, ASAT e ALAT não afetaram estatisticamente significativamente a farmacocinética de 5 "-DFUR, 5-FU e FBAL.
Pacientes com função hepática prejudicada devido a metástases hepáticas: Um estudo farmacocinético demonstrou que a biodisponibilidade da capecitabina e a exposição ao 5-FU podem ser aumentadas em pacientes com câncer com insuficiência hepática leve a moderada devido a metástases hepáticas em comparação com pacientes sem insuficiência hepática disponibilidade de dados farmacocinéticos em pacientes com insuficiência hepática grave. .
Pacientes com insuficiência renalCom base nos resultados de um estudo farmacocinético realizado em pacientes com câncer com insuficiência renal leve a grave, não há evidências de que a depuração da creatinina afete a farmacocinética do fármaco original e do 5-FU. Verificou-se que a depuração da creatinina influencia a "exposição sistêmica a 5" -DFUR (aumento de 35% na AUC quando a depuração da creatinina diminui em 50%) e FBAL (aumento de 114% na AUC quando a depuração da creatinina diminui em 50%). 50%). O FBAL é um metabólito sem atividade antiproliferativa.
Cidadãos idosos: Com base em análises farmacocinéticas populacionais realizadas em pacientes de várias idades (27 a 86 anos) e dos quais 234 (46%) pacientes tinham 65 anos de idade ou mais, a idade não afeta a farmacocinética de 5 "-DFUR e 5-FU . A AUC de FBAL aumentou com a idade (um aumento de 20% na idade leva a um aumento de 15% na AUC de FBAL). Este aumento é provavelmente devido a uma alteração na função renal.
Fatores étnicos: Após a administração oral de capecitabina 825 mg / m2 duas vezes ao dia durante 14 dias, os pacientes japoneses (n = 18) tiveram aproximadamente 36% menor Cmax e 24% menor AUC para capecitabina em comparação com pacientes caucasianos (n = 22). Os pacientes japoneses também tiveram aproximadamente 25% menor Cmax e 34% menor AUC para FBAL do que pacientes caucasianos. A relevância clínica dessas diferenças é desconhecida. Não houve diferenças significativas na exposição a outros metabólitos (5 "DFCR, 5" DFUR e 5-FU).
05.3 Dados de segurança pré-clínica
Em estudos de toxicidade de dose repetida, a administração oral diária de capecitabina a macacos cynomolgus e camundongos produziu efeitos tóxicos gastrointestinais, hematopoiéticos e linfáticos típicos das fluoropirimidinas. Essas toxicidades eram reversíveis. Foi observada toxicidade cutânea, caracterizada por alterações degenerativas / regressivas, devido à capecitabina. A capecitabina não mostrou evidência de toxicidade hepática e do SNC. A toxicidade cardiovascular (por exemplo, prolongamento do intervalo PR e intervalo QT) foi identificada no macaco cynomolgus após administração intravenosa (100 mg / kg), mas não após dosagem repetida (1379 mg / m2 / dia). Por via oral.
Um estudo de carcinogenicidade de dois anos em camundongos não produziu evidências de carcinogenicidade devido à capecitabina.
Em estudos de fertilidade padrão, ratos fêmeas tomando capecitabina exibiram distúrbios de fertilidade; este efeito foi, no entanto, reversível após um período de suspensão do medicamento. Além disso, alterações atróficas e degenerativas foram encontradas nos órgãos reprodutivos de camundongos machos durante um estudo de 13 semanas; no entanto, esses efeitos foram reversíveis após um período de retirada do medicamento (ver seção 4.6).
Estudos sobre embriotoxicidade e teratogenicidade em camundongos mostraram um aumento relacionado à dose na reabsorção fetal e na teratogenicidade. No macaco, abortos e letalidade embrionária foram observados em altas doses, mas não houve evidência de teratogenicidade.
Capecitabina não foi mutagênica em vitro para bactérias (teste de Ames) ou para células de mamíferos (teste de mutação do gene V79 / HPRT do hamster chinês). No entanto, como outros análogos de nucleosídeos (ou seja, 5-FU), a capecitabina era clastogênica em linfócitos humanos (em vitro) e mostrou uma tendência positiva no teste (na Vivo) de micronúcleos na medula óssea de camundongo.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Núcleo do tablet:
lactose anidra,
croscarmelose de sódio,
hipromelose,
celulose microcristalina,
estearato de magnesio.
Revestimento de comprimido:
hipromelose,
dióxido de titânio (E171),
óxido de ferro amarelo e vermelho (E172),
talco.
06.2 Incompatibilidade
Não é relevante.
06.3 Período de validade
3 anos.
06.4 Precauções especiais para armazenamento
Não armazene acima de 30 ° C.
06.5 Natureza da embalagem primária e conteúdo da embalagem
Natureza: bolha de PVC / PVDC.
Conteúdo: 60 comprimidos revestidos por película (6 blisters de 10 comprimidos).
06.6 Instruções de uso e manuseio
Sem instruções especiais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
Reino Unido
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU / 1/00/163/001
035219017
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 2 de fevereiro de 2001
Data da última renovação: 2 de fevereiro de 2006
10.0 DATA DE REVISÃO DO TEXTO
Setembro 2015











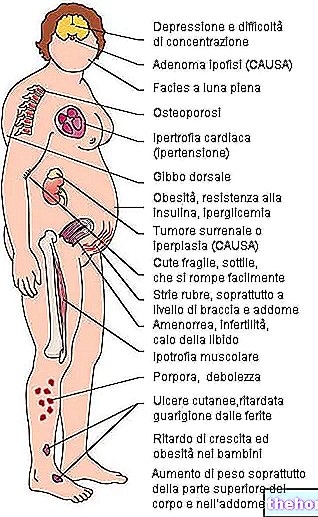





.jpg)