Generalidade
O retinoblastoma (Rb) é um tumor maligno do olho que se desenvolve a partir das células da retina. Esse câncer pode ocorrer em qualquer idade, mas o início é mais comum durante a infância, antes dos cinco anos.

O câncer infantil é agressivo: o retinoblastoma pode se espalhar para os gânglios linfáticos, ossos ou medula óssea. Raramente, envolve o sistema nervoso central (cérebro e medula espinhal).
Cerca de 90% das crianças com retinoblastoma têm prognóstico positivo (probabilidade de cura), desde que o diagnóstico seja precoce e o tratamento seja iniciado antes que o câncer se espalhe. Sempre que possível, o objetivo da intervenção médica é preservar a visão do paciente.
Causas
A série de eventos que levam ao aparecimento do tumor é complexa e começa quando as células da retina desenvolvem uma mutação (ou deleção), envolvendo o gene supressor de tumor RB1, localizado na banda q14 do cromossomo 13 (13q14).
Cada célula normalmente possui dois genes RB1:
- Se pelo menos uma cópia do gene funcionar corretamente, o retinoblastoma não surge (mas o risco aumenta);
- Quando ambas as cópias do gene sofrem mutação ou estão ausentes, ocorre a proliferação celular descontrolada.
Em muitos casos, não está claro o que exatamente induz mudanças no gene RB1 (retinoblastoma esporádico); estes podem resultar de erros genéticos aleatórios, que ocorrem, por exemplo, durante a reprodução e divisão celular. No entanto, sabe-se que as anormalidades genéticas subjacentes ao retinoblastoma também podem ser transmitidas de pais para filhos, com um padrão de herança autossômico dominante. Isso significa que se um pai carrega um gene mutado (dominante), cada filho terá 50% de chance de herdá-lo e 50% de chance de ter uma composição genética normal (genes recessivos).
- Uma célula ocasional inativa sua única cópia normal do gene RB1 (uma cópia já está mutada);
- A perda das duas cópias de RB1 leva a uma "proliferação excessiva da retina.
- Uma célula ocasional inativa um de seus genes RB1 normais;
- A segunda cópia do gene RB1 está inativada;
- A perda das duas cópias de RB1 induz uma proliferação celular excessiva que leva ao retinoblastoma.
Características genéticas e moleculares
- O retinoblastoma foi o primeiro tumor diretamente associado a uma "anormalidade genética (deleção ou mutação da banda q14 do cromossomo 13).
- O RB1 codifica a proteína pRb, que desempenha um papel fundamental no ciclo celular: permite a replicação do DNA e a progressão do ciclo celular, pois participa do controle da transcrição dos genes da fase S (G1 → † "S).
- Além do retinoblastoma, o gene RB1 está inativado nos cânceres de bexiga, mama e pulmão.
Retinoblastoma hereditário
Crianças com retinoblastoma hereditário tendem a desenvolver a doença mais cedo do que os casos esporádicos. Além disso, essas crianças apresentam risco aumentado para outros cânceres não oculares, pois a anormalidade no gene RB1 é congênita (ou seja, presente desde o nascimento) e afeta todas as células do corpo (conhecida como mutação da linha germinativa), incluindo as de ambos. retinas: por esse motivo, as crianças com a forma hereditária geralmente apresentam retinoblastoma bilateral, em vez de apenas um olho.
Sintomas
Para saber mais: Sintomas de retinoblastoma
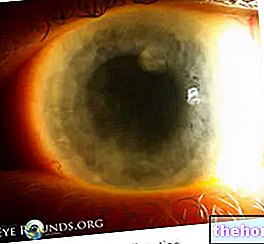
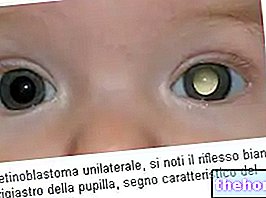
O sinal mais comum e óbvio de retinoblastoma é a aparência anormal da pupila, que apresenta um reflexo branco-acinzentado quando é atingida por um feixe de luz (leucocoria ou reflexo amaurótico de gato). Outros sinais e sintomas incluem: diminuição da visão, dor e vermelhidão nos olhos e atraso no desenvolvimento. Algumas crianças com retinoblastoma podem desenvolver estrabismo (olhos desalinhados); em outros casos, é possível encontrar glaucoma neovascular, que, depois de algum tempo, pode causar aumento do olho (buftalmo).
As células cancerosas podem invadir ainda mais o olho e outras estruturas:
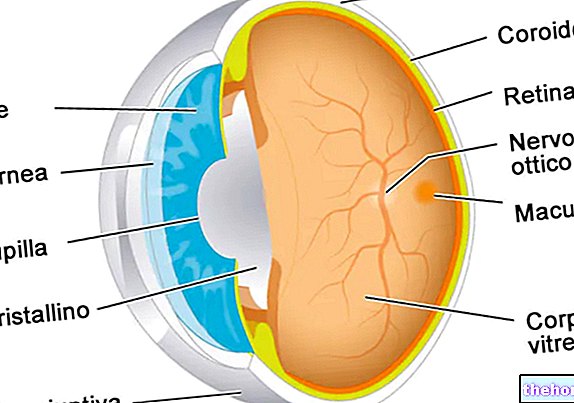
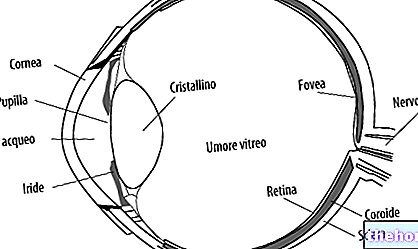
- Retinoblastoma intraocular. O retinoblastoma pode ser definido como intraocular quando o tumor está inteiramente localizado dentro do olho. A neoplasia pode ser encontrada apenas na retina ou também afetar outras partes, como a coróide, corpo ciliar e parte do nervo óptico. O retinoblastoma intraocular, portanto, não se espalha para os tecidos ao redor do olho.
- Retinoblastoma extraocular. O tumor pode proliferar para afetar os tecidos ao redor do olho (retinoblastoma orbital) O câncer também pode se espalhar para outras áreas do corpo, como cérebro, coluna, medula óssea e gânglios linfáticos (retinoblastoma metastático).
A presença de extensão orbital, envolvimento uveal e invasão do nervo óptico são fatores de risco conhecidos para o desenvolvimento de retinoblastoma metastático.
Diagnóstico
Em caso de história familiar positiva, o paciente é submetido a exames oftalmológicos regulares para rastreamento do câncer. Se o retinoblastoma congênito for bilateral, geralmente é diagnosticado no primeiro ano de vida, enquanto quando afeta apenas um olho, a presença do tumor pode ser confirmada por volta dos 18-30 meses de idade.
O diagnóstico clínico do retinoblastoma é feito pelo exame do fundo, podendo o tumor, dependendo da localização, ser visível ao simples exame do olho, por meio da oftalmoscopia indireta. As técnicas de imagem podem ser usadas para confirmar o diagnóstico, definir o estadiamento do tumor (onde está, quão difundido está, se está afetando as funções de outros órgãos do corpo, etc.) e determinar se o tratamento foi eficaz . As investigações podem incluir ultrassom, tomografia computadorizada (TC) e ressonância magnética (MRI).
O diagnóstico genético-molecular é possível através da identificação da mutação do gene RB1. A análise citogenética (ou seja, dos cromossomos) dos linfócitos do sangue periférico é usada para detectar deleções ou rearranjos envolvendo o cromossomo 13 (13q14.1-q14. 2) .
Tratamentos
No caso do retinoblastoma, várias opções de tratamento podem ser utilizadas.
Os objetivos do tratamento são:
- Elimine o tumor e salve a vida do paciente;
- Salve o olho, se possível;
- Preserve a visão tanto quanto possível;
- Evite o desenvolvimento de outros tipos de câncer, que também podem ser causados pelo tratamento, especialmente em crianças com retinoblastoma hereditário.
O prognóstico (probabilidade de recuperação) e as opções de tratamento dependem dos seguintes fatores:
- Estágio do tumor;
- Idade do paciente e estado geral de saúde;
- Localização, tamanho e número de focos tumorais;
- Propagação do câncer para outras áreas além do globo ocular
- Quão provável é que a visão possa ser preservada em um ou em ambos os olhos.
A maioria dos casos de retinoblastoma é diagnosticada precocemente e tratada com sucesso, antes que o câncer possa metastatizar para fora do globo ocular, resultando em uma taxa de cura de mais de 90%.