Ingredientes ativos: Pegfilgrastim
Neulasta 6 mg solução injetável em seringa pré-cheia
Indicações Por que o Neulasta é usado? Para que serve?
O Neulasta contém a substância ativa pegfilgrastim. Pegfilgrastim é uma proteína produzida por uma técnica biotecnológica em uma célula bacteriana chamada Escherichia coli. Pertence a um grupo de proteínas chamadas citocinas e é muito semelhante a uma proteína natural (fator estimulador de colônias de granulócitos) produzida pelo nosso corpo.
Neulasta é utilizado para reduzir a duração da neutropenia (contagem baixa de glóbulos brancos) e a ocorrência de neutropenia febril (contagem baixa de glóbulos brancos com febre) que pode ser causada por quimioterapia citotóxica (medicamentos que destroem células de crescimento rápido). Os glóbulos brancos são importantes porque ajudam o corpo a combater infecções. Essas células são muito sensíveis aos efeitos da quimioterapia, que podem causar uma diminuição no número dessas células no corpo. Se a contagem de seus glóbulos brancos cair para um nível baixo, pode não haver quantidade suficiente para combater a bactéria e você corre um risco maior de contrair uma infecção.
O seu médico receitou Neulasta para estimular a sua medula óssea (a parte do osso que produz as células sanguíneas) a produzir mais glóbulos brancos para ajudar o seu corpo a combater infecções.
Contra-indicações Quando Neulasta não deve ser usado
Não use Neulasta se você é alérgico a pegfilgrastim, filgrastim, proteínas derivadas de Escherichia coli ou qualquer outro componente deste medicamento.
Precauções de uso O que você precisa saber antes de tomar Neulasta
Fale com o seu médico ou farmacêutico ou enfermeiro antes de usar Neulasta:
- se tiver uma reação alérgica incluindo fraqueza, queda da pressão arterial, dificuldade em respirar, inchaço da face (anafilaxia), vermelhidão e vermelhidão, erupção cutânea e áreas da pele com comichão
- se tem alergia ao látex. A tampa da agulha da seringa pré-cheia contém um derivado do látex que pode causar reações alérgicas graves
- se tem tosse, febre e dificuldade em respirar. Isso pode ser um sinal de Síndrome da Dificuldade Respiratória Aguda (SDRA).
- se você tiver um ou mais dos seguintes efeitos colaterais:
- inchaço ou inchaço, que pode estar associado a menos passagem de fluidos, dificuldade em respirar, inchaço abdominal e uma sensação de plenitude e uma sensação geral de cansaço. Estes podem ser sintomas de uma condição chamada "Síndrome de Vazamento Capilar", causando perfusão de sangue de pequenos vasos no corpo. Veja o parágrafo 4.
- se tem dor na parte superior esquerda do abdómen ou dor na extremidade do ombro. Estes podem ser sinais de um problema no baço (esplenomegalia)
- se você teve recentemente uma "infecção pulmonar grave (pneumonia), fluido nos pulmões (edema pulmonar), inflamação dos pulmões (doença pulmonar intersticial) ou uma" anormalidade encontrada em raios-X (infiltração pulmonar)
- se sabe que tem contagens de células sanguíneas anormais (por exemplo, aumento dos glóbulos brancos ou anemia) ou uma diminuição dos níveis de plaquetas, o que reduz a capacidade de coagulação do corpo (trombocitopenia). Seu médico pode querer monitorá-lo de perto
- se tem anemia falciforme. Seu médico pode querer monitorá-lo de perto
- se tiver repentinamente sinais de alergia, como erupção na pele, urticária ou comichão na pele, inchaço da face, lábios, língua ou outras partes do corpo, falta de ar, respiração ruidosa ou dificuldade em respirar, estes podem ser sinais de uma reacção alérgica grave.
O seu médico irá verificar o seu sangue e urina regularmente, uma vez que Neulasta pode danificar os minúsculos filtros no interior dos rins (glomerulonefrite).
Você deve conversar com seu médico sobre os riscos de desenvolver câncer no sangue. Se tem ou pode ter cancro do sangue, não deve utilizar Neulasta, a menos que o seu médico lhe diga para o fazer.
Perda de resposta ao pegfilgrastim
Se você diminuiu a resposta ou não manteve a resposta ao tratamento com pegfilgrastim, o seu médico investigará as razões, incluindo a possibilidade de ter desenvolvido anticorpos que neutralizam a atividade do pegfilgrastim.
Interações Quais medicamentos ou alimentos podem modificar o efeito do Neulasta
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Avisos É importante saber que:
Gravidez e amamentação
Consulte o seu médico ou farmacêutico antes de tomar qualquer medicamento. Neulasta não foi testado em mulheres grávidas. É importante informar o seu médico se:
- vocÊ esta grávida;
- suspeita de gravidez; ou
- está planejando uma gravidez.
Se engravidar durante o tratamento com Neulasta, informe o seu médico. Você pode ser incentivado a se inscrever no Programa de Vigilância de Gravidez da Amgen. Os detalhes de contato do representante local são fornecidos na seção 6 deste folheto.
A menos que seu médico lhe diga o contrário, você deve interromper a amamentação se usar Neulasta.
Se está a amamentar enquanto está a tomar Neulasta, pode ser encorajada a inscrever-se no programa de Vigilância da Lactação da Amgen.Os contactos do seu representante local são fornecidos na secção 6 deste folheto.
Condução e utilização de máquinas
Neulasta não tem ou tem uma influência negligenciável sobre a capacidade de conduzir ou utilizar máquinas.
Neulasta contém sorbitol (E420) e acetato de sódio
Neulasta contém sorbitol (um tipo de açúcar). Se foi informado pelo seu médico que tem "intolerância a alguns açúcares, contacte-o antes de tomar este medicamento.
Este medicamento contém menos de 1 mmol (23 mg) de sódio por cada dose de 6 mg, é essencialmente isento de sódio.
Dose, Método e Tempo de Administração Como usar Neulasta: Posologia
Neulasta é indicado em adultos com 18 ou mais anos.
Tome Neulasta sempre de acordo com as indicações do médico. Em caso de dúvida, consulte o seu médico ou farmacêutico. A dose usual é uma injeção subcutânea (injeção sob a pele) de 6 mg usando uma seringa pré-cheia, que deve ser administrada pelo menos 24 horas após a última dose de quimioterapia no final de cada ciclo de quimioterapia.
Não agite o Neulasta vigorosamente, pois pode comprometer a sua atividade.
Como se injetar Neulasta
O seu médico pode considerar que é melhor para si injectar Neulasta a si próprio.O seu médico ou enfermeiro mostrar-lhe-á como injectar Neulasta.Não tente injectar-se se não lhe foi dito como injectar.
Leia a secção no final deste folheto para obter instruções sobre como injectar Neulasta a si próprio.
Se você esquecer sua injeção de Neulasta
Se se esqueceu de uma dose de Neulasta, deve contactar o seu médico para determinar quando deve ser administrada a próxima injeção.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
Sobredosagem O que fazer se você tiver tomado Neulasta em demasia
Se utilizar mais Neulasta do que deveria, deve contactar o seu médico, farmacêutico ou enfermeiro.
Efeitos colaterais Quais são os efeitos colaterais do Neulasta
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
Informe o seu médico imediatamente se sentir algum ou uma combinação dos seguintes efeitos colaterais:
- inchaço ou inchaço, que pode estar associado à passagem de água com menos frequência, dificuldade em respirar, inchaço e sensação de plenitude e uma sensação geral de cansaço. Esses sintomas geralmente se desenvolvem rapidamente.
Estes podem ser sintomas de uma condição incomum (pode afetar até 1 em 100 pessoas) chamada "síndrome de vazamento capilar", que faz com que o sangue vaze de pequenos vasos sanguíneos para o corpo e requer atenção médica urgente.
Efeitos colaterais muito comuns (que podem afetar mais de 1 em 10 pessoas):
- dor no osso. O seu médico irá dizer-lhe o que deve tomar para aliviar as dores nos ossos.
- náusea e dor de cabeça.
Efeitos colaterais comuns (que podem afetar até 1 em cada 10 pessoas):
- dor no local da injeção.
- dores gerais e dores nas articulações e nos músculos.
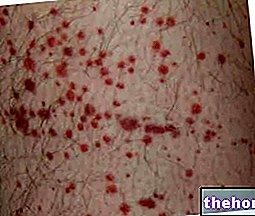
- algumas alterações podem ocorrer no sangue, mas serão detectadas durante os exames de sangue de rotina. Os níveis de glóbulos brancos podem aumentar por um curto período de tempo. Os níveis de plaquetas podem cair causando hematomas.
Efeitos colaterais incomuns (que podem afetar até 1 em 100 pessoas):
- reações do tipo alérgico, incluindo vermelhidão e rubor, erupção cutânea (vermelhidão da pele) e inchaço da pele com comichão.
- reações alérgicas graves, incluindo anafilaxia (fraqueza, queda da pressão arterial, dificuldade em respirar, inchaço da face).
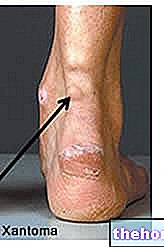
- aumento do volume do baço.
- ruptura do baço. Alguns casos de ruptura do baço foram fatais. É importante que contacte o seu médico imediatamente se sentir dor na parte superior esquerda do abdómen ou no ombro esquerdo, pois pode indicar problemas com o baço.
- problemas respiratórios. Se tiver tosse, febre e dificuldade em respirar, contacte o seu médico.
- houve casos de síndrome de Sweet (lesões roxas, elevadas e dolorosas nos membros e às vezes no rosto e pescoço, associadas à febre), mas outros fatores podem ter contribuído
- vasculite cutânea (inflamação dos vasos sanguíneos da pele).
- danos aos minúsculos filtros dentro dos rins (glomerulonefrite).
- vermelhidão no local da injeção.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico ou enfermeiro. Isto inclui quaisquer efeitos secundários possíveis não listados neste folheto. Também pode comunicar os efeitos secundários diretamente através do sistema nacional de notificação listado no Apêndice V.
Expiração e retenção
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no rótulo da seringa após VAL. O prazo de validade corresponde ao último dia desse mês.
Conservar no frigorífico (2 ° C - 8 ° C).
Pode retirar o Neulasta do frigorífico e mantê-lo à temperatura ambiente (não superior a 30 ° C) durante não mais do que 3 dias. Assim que a seringa for retirada do frigorífico e atingir a temperatura ambiente (não superior a 30 ° C), deve ser utilizada no prazo de 3 dias ou rejeitada.
Não congele. Neulasta pode ser utilizado se tiver sido acidentalmente congelado uma vez durante menos de 24 horas.
Manter o recipiente dentro da embalagem exterior para proteger o medicamento da luz.
Não use este medicamento se notar que está turvo ou se observar partículas.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Prazo "> Outras informações
O que Neulasta contém
- O ingrediente ativo é pegfilgrastim. Cada seringa pré-cheia contém 6 mg de pegfilgrastim em 0,6 ml de solução.
- Os outros componentes são acetato de sódio, sorbitol (E420), polissorbato 20 e água para preparações injetáveis. Veja o parágrafo 2.
Qual a aparência de Neulasta e conteúdo da embalagem
Neulasta é uma solução injetável límpida e incolor em seringa pré-cheia (6 mg / 0,6 ml).
Cada embalagem contém 1 seringa pré-cheia de vidro tipo I. Com agulha de aço inoxidável e tampa da agulha As seringas são embaladas com blister ou sem blister.
Validade "> Instruções para a injeção da seringa pré-cheia de Neulasta
Esta seção contém informações sobre como injetar Neulasta em si mesmo.
É importante que não tente injetar-se se não tiver sido informado pelo seu médico, enfermeiro ou farmacêutico como o deve fazer. Se tiver dúvidas sobre como injetar, peça ajuda ao seu médico, enfermeiro ou farmacêutico.
Como utilizar, por si ou pela pessoa que lhe administrou a injeção, de Neulasta em seringa pré-cheia
Terá de se injectar imediatamente por baixo da pele.Esta injecção denomina-se subcutânea.
O que é preciso
Para administrar a si mesmo uma "injeção subcutânea, você precisará de:
- uma seringa pré-cheia de Neulasta; E
- lenços com álcool ou desinfetantes semelhantes.
O que devo fazer antes de dar a mim mesmo uma "injeção subcutânea de Neulasta?
- Tire o remédio da geladeira.
- Não agite a seringa pré-cheia.
- Não retire a tampa da agulha da seringa até estar pronto para injetar.
- Verifique o prazo de validade no rótulo da seringa pré-cheia (VAL). Não use após o último dia do mês indicado.
- Verifique o aspecto do Neulasta. Deve ser um líquido límpido e incolor. Se vir partículas, não deve utilizá-lo.
- Para uma injeção mais confortável, deixe a seringa pré-cheia fora do refrigerador por meia hora para atingir a temperatura ambiente ou segure-a suavemente em sua mão por alguns minutos. Não aqueça o Neulasta de nenhuma outra forma (por exemplo, não aqueça em um forno de micro-ondas ou água quente).
- Lave bem as mãos.
- Encontre uma superfície confortável, bem iluminada e limpa e tenha tudo o que você precisa à mão.
Como preparo a injeção de Neulasta?
Antes de administrar a injeção de Neulasta a si próprio deve fazer o seguinte:
- Pegue a seringa nas mãos e retire com cuidado a tampa da agulha sem dobrá-la. Puxe horizontalmente. Não toque na agulha nem empurre o êmbolo.
- Poderá notar uma pequena bolha de ar na seringa pré-cheia. Não deve remover a bolha de ar antes de injetar. Injetar a solução com a bolha de ar é inofensivo.
- Agora você pode usar a seringa pré-cheia.
Onde devo aplicar a injeção em mim mesmo?
Os locais mais adequados para se injetar são:
- a parte superior das coxas; E
- o abdômen, exceto a área ao redor do umbigo.
Se outra pessoa aplicar a injeção, você também poderá usar a parte de trás dos braços.
Como faço para me dar a injeção?
- Limpe a pele com algodão embebido em álcool.
- Levante a pele entre o polegar e o indicador (sem apertar). Enfie a agulha na pele.
- Empurre o êmbolo para baixo com uma pressão lenta e constante. Empurre o êmbolo totalmente para dentro até que todo o líquido tenha sido injetado.
- Após injetar o líquido, retire a agulha e solte a pele.
- Se notar uma pequena gota de sangue no local da injeção, limpe-a suavemente com um cotonete ou gaze. Não esfregue o local da injeção. Se necessário, você pode cobrir o local da injeção com um adesivo.
- Não reutilize o Neulasta restante na seringa.
Lembrar
Use cada seringa apenas para uma injeção. Se tiver qualquer problema, não hesite em consultar o seu médico ou enfermeiro para obter ajuda e aconselhamento.
Eliminação de seringas usadas
- Não coloque a tampa de volta em agulhas usadas.
- Mantenha as seringas usadas fora da vista e do alcance das crianças.
- As seringas usadas devem ser eliminadas de acordo com os requisitos locais. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isso ajudará a proteger o meio ambiente.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO -
NEULASTA 6 MG SOLUÇÃO PARA INJEÇÃO
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA -
Cada seringa pré-cheia contém 6 mg de pegfilgrastim * em 0,6 ml de solução injetável. A concentração é de 10 mg / ml considerando apenas a porção proteica **.
* Pegfilgrastim é produzido em células de Escherichia coli com tecnologia de DNA recombinante e subsequente conjugação com polietilenoglicol (PEG).
** A concentração é de 20 mg / ml se a porção PEG da molécula for incluída.
A potência deste produto não deve ser comparada com a de qualquer outra proteína peguilada ou não peguilada pertencente à mesma classe terapêutica.
Para obter mais informações, consulte a seção 5.1.
Excipiente (s) com efeito conhecido:
Cada seringa pré-cheia contém 30 mg de sorbitol (E420)
Cada seringa pré-cheia contém menos de 1 mmol (23 mg) de sódio (ver secção 4.4).
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA -
Solução injetável.
Solução injetável límpida e incolor.
04.0 INFORMAÇÕES CLÍNICAS -
04.1 Indicações terapêuticas -
Redução da duração da neutropenia e da incidência de neutropenia febril em pacientes adultos tratados com quimioterapia citotóxica para câncer (com exceção da leucemia mielóide crônica e síndromes mielodisplásicas).
04.2 Posologia e método de administração -
A terapia com Neulasta deve ser iniciada e supervisionada por médicos com experiência em oncologia e / ou hematologia.
Dosagem
Recomenda-se uma dose de 6 mg (uma única seringa pré-cheia) de Neulasta para cada ciclo de quimioterapia, administrada pelo menos 24 horas após a quimioterapia citotóxica.
Método de administração
Neulasta é injetado por via subcutânea. A injeção deve ser administrada na coxa, abdómen ou braço. Para obter instruções sobre o manuseamento do medicamento antes da administração, ver secção 6.6.
População pediátrica
A segurança e eficácia de Neulasta em crianças não foram ainda estabelecidas Os dados atualmente disponíveis estão descritos nas secções 4.8, 5.1 e 5.2, mas não pode ser feita nenhuma recomendação quanto à posologia.
Pacientes com insuficiência renal
Nenhum ajuste de dose é recomendado em pacientes com insuficiência renal, incluindo aqueles com doença renal em estágio terminal.
04.3 Contra-indicações -
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes.
04.4 Advertências especiais e precauções adequadas de uso -
Dados clínicos limitados sugerem um efeito comparável do pegfilgrastim em comparação com o filgrastim no tempo para a remissão da neutropenia grave em pacientes com leucemia mielóide aguda. de novo (consulte a seção 5.1). No entanto, os efeitos a longo prazo do Neulasta na leucemia mieloide aguda não foram estabelecidos; portanto, o produto deve ser usado com cautela nesta população de pacientes.
O fator estimulador de colônias de granulócitos pode promover o crescimento de células mieloides em vitro e efeitos semelhantes podem ser observados em vitro em algumas células não mieloides.
A segurança e eficácia de Neulasta não foram estudadas em pacientes com síndrome mielodisplásica, leucemia mielóide crônica e em pacientes com leucemia mielóide aguda secundária (LMA); portanto, não deve ser usado nesses pacientes. Deve-se ter cuidado especial para distinguir o diagnóstico de transformação blástica de leucemia mieloide crônica daquela de leucemia mieloide aguda.
A eficácia e segurança da administração de Neulasta em pacientes com LMA de novo de idade
Não foram estudadas a segurança e eficácia de Neulasta em doentes a receber quimioterapia em altas doses.Este medicamento não deve ser utilizado para aumentar as doses de quimioterapia citotóxica para além dos regimes posológicos padrão.
Eventos adversos pulmonares
Após a administração de G-CSF, foram notificadas reações adversas pulmonares pouco frequentes (≥ 1 / 1.000, pneumonia intersticial. Os doentes com história recente de infiltrados pulmonares ou pneumonia podem estar em maior risco (ver secção 4.8).
O início de sintomas pulmonares, como tosse, febre e dispneia, ao mesmo tempo que um quadro radiológico de infiltrados pulmonares e uma deterioração da função pulmonar, associado a uma contagem elevada de leucócitos, pode ser os sinais iniciais da síndrome da angústia respiratória aguda (Síndrome respiratória aguda Grave, ARDS). Nessas circunstâncias, ao critério do médico, a terapêutica com Neulasta deve ser descontinuada e instituído um tratamento adequado (ver secção 4.8).
Glomerulonefrite
Glomerulonefrite foi relatada em pacientes que receberam filgrastim e pegfilgrastim. Geralmente, os eventos de glomerolunefrite se resolvem após a redução da dose ou descontinuação de filgrastim e pegfilgrastim. O monitoramento de urinálise é recomendado.
Síndrome de vazamento capilar
A síndrome de vazamento capilar foi relatada após a administração de fatores estimuladores de colônias de granulócitos e é caracterizada por hipotensão, hipoalbuminemia, edema e hemoconcentração. Os doentes que desenvolvem sintomas de síndrome de fuga capilar devem ser cuidadosamente monitorizados e receber tratamento sintomático padrão, que pode incluir a necessidade de cuidados intensivos (ver secção 4.8).
Esplenomegalia e ruptura esplênica
Após a administração de pegfilgrastim, foram notificados casos pouco frequentes, mas geralmente assintomáticos, de esplenomegalia e casos pouco frequentes de ruptura esplénica, incluindo alguns casos fatais (ver secção 4.8). Portanto, o volume do baço deve ser monitorado cuidadosamente (por exemplo, por exame clínico, ultrassom). Um diagnóstico de ruptura esplênica deve ser considerado em pacientes que apresentam dor abdominal no quadrante superior esquerdo ou dor no ombro.
Trombocitopenia e anemia
O tratamento com Neulasta sozinho não impede a trombocitopenia e a anemia causadas pela manutenção de doses completas de quimioterapia mielossupressora conforme programado. Recomenda-se o monitoramento regular da contagem de plaquetas e do hematócrito. Deve-se prestar atenção especial ao administrar agentes quimioterápicos únicos ou combinados que causam trombocitopenia grave.
Anemia falciforme
As crises de células falciformes foram associadas ao uso de pegfilgrastim em pacientes com traço falciforme ou com doença falciforme (ver seção 4.8). Portanto, o médico deve ter cautela ao prescrever Neulasta a pacientes com traço falciforme ou doença falciforme. mantenha os parâmetros clínicos e laboratoriais adequados e deve prestar atenção à possível associação entre este medicamento e um baço aumentado e uma crise vaso-oclusiva.
Leucocitose
Valores de glóbulos brancos (Glóbulo branco, Leucócitos) igual ou superior a 100 x 109 / l foram observados em menos de 1% dos pacientes tratados com Neulasta. Não foram relatados eventos adversos diretamente atribuíveis a este grau de leucocitose. Este aumento na contagem de leucócitos é transitório , é tipicamente observado 24-48 horas após a administração e é consistente com os efeitos farmacodinâmicos deste medicamento. Consistente com os efeitos clínicos e a possibilidade de leucocitose, uma contagem de leucócitos (leucócitos) deve ser realizada em intervalos regulares durante a terapia. a contagem de glóbulos brancos excede 50 x 109 / L após o nadir esperado, a administração deste medicamento deve ser interrompida imediatamente.
Hipersensibilidade
Foram notificadas reações de hipersensibilidade, incluindo reações anafiláticas, que ocorrem no início ou após o tratamento em doentes tratados com Neulasta. Descontinue definitivamente o tratamento com Neulasta em doentes com hipersensibilidade clinicamente significativa. Não administre Neulasta a doentes com história de hipersensibilidade ao pegfilgrastim ou filgrastim Se ocorrer uma reação alérgica grave, deve ser administrada terapia apropriada, seguida de acompanhamento cuidadoso do paciente por vários dias.
Imunogenicidade
Tal como acontece com todas as proteínas terapêuticas, existe um risco potencial de imunogenicidade. A probabilidade de gerar anticorpos contra o pegfilgrastim é geralmente baixa. O desenvolvimento de anticorpos de ligação é esperado com todos os produtos biológicos; no entanto, até à data, não foram associados à atividade. Neutralizante.
A segurança e eficácia de Neulasta na mobilização de células progenitoras hematopoiéticas em pacientes saudáveis ou dadores não foram avaliadas de forma adequada.
A tampa da agulha da seringa pré-cheia contém borracha natural seca (um derivado do látex) que pode causar reações alérgicas.
O aumento da atividade hematopoiética da medula óssea em resposta à terapia com fator de crescimento foi associado a achados radiológicos ósseos positivos transitórios, o que deve ser considerado na interpretação dos dados radiológicos.
Neulasta contém sorbitol. Os doentes com problemas hereditários raros de intolerância à frutose não devem tomar este medicamento.
Neulasta contém menos de 1 mmol (23 mg) de sódio numa dose de 6 mg, ou seja, é praticamente “isento de sódio”.
Para melhorar a rastreabilidade dos fatores estimuladores de colônias de granulócitos (G-CSF), o nome comercial do produto administrado deve ser claramente registrado no prontuário do paciente.
04.5 Interações com outros medicamentos e outras formas de interação -
Dada a potencial sensibilidade das células mieloides em divisão rápida à quimioterapia citotóxica, Neulasta deve ser administrado pelo menos 24 horas após a administração da quimioterapia citotóxica. Em estudos clínicos, a administração de Neulasta 14 dias antes da quimioterapia demonstrou ser segura. A utilização de Neulasta concomitantemente com qualquer quimioterapia não foi avaliada em doentes.Em modelos animais, a administração concomitante de Neulasta e 5-fluorouracilo (5-FU) ou outros antimetabólitos demonstrou agravar a mielossupressão..
Os estudos clínicos não investigaram especificamente possíveis interações com outros fatores de crescimento hematopoiéticos e citocinas.
A potencial interação com o lítio, que também promove a liberação de neutrófilos, não foi especificamente estudada, não havendo evidências de que essa interação possa ser prejudicial.
A segurança e eficácia de Neulasta não foram avaliadas em pacientes recebendo quimioterapia associada a mielossupressão retardada, como nitrosoureias.
Não foram realizados estudos específicos sobre interações ou metabolismo; no entanto, os estudos clínicos não mostraram quaisquer interações de Neulasta com outros medicamentos.
04.6 Gravidez e amamentação -
Gravidez
Não existem dados ou são limitados os dados sobre a utilização de pegfilgrastim em mulheres grávidas Os estudos em animais revelaram toxicidade reprodutiva (ver secção 5.3) .Neulasta não é recomendado durante a gravidez e em mulheres com potencial para engravidar que não utilizam métodos contraceptivos.
Mulheres grávidas durante o tratamento com Neulasta são incentivadas a se inscrever no Programa de Vigilância de Gravidez da Amgen. Os detalhes de contato são fornecidos na seção 6 do Folheto Informativo.
Hora da alimentação
A informação sobre a excreção de Neulasta / metabolitos no leite humano é insuficiente. Não pode ser excluído um risco para os recém-nascidos / lactentes. Deve ser tomada uma decisão sobre descontinuar a amamentação ou descontinuar / abster-se da terapêutica com Neulasta tendo em conta o benefício da amamentação para o bebê e o benefício da terapia para a mulher.
As mulheres que amamentam durante o tratamento com Neulasta são incentivadas a se inscrever no Programa de Vigilância da Lactação da Amgen. Os detalhes de contato são fornecidos na seção 6 do Folheto Informativo.
Fertilidade
Pegfilgrastim não teve efeito sobre o desempenho reprodutivo ou fertilidade em ratos machos ou fêmeas com a dose semanal cumulativa de aproximadamente 6 a 9 vezes a dose humana recomendada mais elevada (com base na área de superfície corporal) (ver secção 5.3).
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas -
Neulasta não tem ou tem uma influência negligenciável sobre a capacidade de conduzir ou utilizar máquinas.
04.8 Efeitos indesejáveis -
Resumo do perfil de segurança
As reações adversas notificadas com mais frequência foram dor óssea (muito frequente [≥ 1/10]) e dor músculo-esquelética (frequente). A dor óssea era geralmente leve a moderada, transitória e controlável com analgésicos comuns na maioria dos pacientes.
Casos de reações de hipersensibilidade, incluindo erupção cutânea, urticária, angioedema, dispneia, eritema, rubor e hipotensão, foram relatados com a primeira ou subseqüentes administrações de Neulasta (incomum [≥ 1 / 1.000, anafilaxia, pode ocorrer em pacientes recebendo Neulasta (incomum) ) (consulte a seção 4.4).
A síndrome de vazamento capilar, que pode ser fatal se o tratamento for adiado, foi relatada como incomum (≥ 1 / 1.000 a
A esplenomegalia, geralmente assintomática, é incomum.
Após a administração de pegfilgrastim, foram notificados casos pouco frequentes de ruptura esplénica, incluindo alguns casos fatais (ver secção 4.4).
Foram notificadas reações adversas pulmonares pouco frequentes, incluindo pneumonia intersticial, edema pulmonar, infiltrados pulmonares e fibrose pulmonar. Casos incomuns resultaram em insuficiência respiratória ou síndrome da dificuldade respiratória aguda (Síndrome respiratória aguda Grave, ARDS) que pode ser fatal (ver secção 4.4).
Foram notificados casos isolados de crises de células falciformes (pouco frequentes nestes doentes) em doentes com traço falciforme ou doença das células falciformes (ver secção 4.4).
Tabela de reações adversas
Os dados da tabela abaixo descrevem as reações adversas notificadas em estudos clínicos e notificações espontâneas. Dentro de cada classe de frequência, os efeitos indesejáveis são relatados em ordem decrescente de gravidade.
¹ Ver seção “Descrição de reações adversas selecionadas” abaixo.
² Esta reação adversa foi identificada através da vigilância pós-comercialização, mas não foi observada em ensaios clínicos randomizados em adultos. A classe de frequência foi determinada com um cálculo estatístico baseado em 1.576 pacientes tratados com Neulasta em nove ensaios clínicos randomizados.
Descrição das reações adversas selecionadas
Foram relatados casos incomuns de síndrome de Sweet, embora a presença subjacente de malignidades hematológicas possa ter contribuído em alguns casos.
Eventos incomuns de vasculite cutânea foram relatados em pacientes tratados com Neulasta. O mecanismo que causa vasculite em pacientes tratados com Neulasta é desconhecido.
Reações no local da injeção, incluindo eritema no local da injeção (incomum (≥ 1 / 1.000,
Foram notificados casos comuns (≥ 1/100, 100 x 109 / l) (ver secção 4.4).
Em doentes tratados com Neulasta após quimioterapia citotóxica, são raras as elevações reversíveis, ligeiras ou moderadas, não acompanhadas de sintomas clínicos, do ácido úrico e da fosfatase alcalina; Elevações reversíveis, leves ou moderadas, não acompanhadas de sintomas clínicos, na desidrogenase láctica são incomuns.
Náusea e dor de cabeça foram observadas muito comumente em pacientes recebendo quimioterapia.
Foram observados casos incomuns de testes de função hepática elevados (LFT) para ALT (alanina aminotransferase) ou AST (aspartato aminotransferase) em pacientes que receberam pegfilgrastim após quimioterapia citotóxica. Esses aumentos são transitórios e reversíveis.
Foram relatados casos comuns de trombocitopenia.
Foram notificados casos de síndrome de fuga capilar após a comercialização com a utilização de fatores estimuladores de colónias de granulócitos.Estes ocorreram geralmente em doentes com doença maligna avançada, sépsis, que estão a tomar vários medicamentos de quimioterapia ou estão em aférese (ver secção 4.4).
População pediátrica
A experiência em crianças é limitada.Uma frequência mais elevada de reações adversas graves foi observada em crianças com 0-5 anos (92%) em comparação com crianças mais velhas com 6-11 e 12-21 anos, respetivamente (80% e 67%) e adultos. O evento adverso mais comum relatado foi dor óssea (ver seções 5.1 e 5.2).
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas ocorridas após a autorização do medicamento é importante porque permite a monitorização contínua da relação benefício / risco do medicamento. Os profissionais de saúde são solicitados a notificar quaisquer suspeitas de reações adversas através do sistema nacional de notificação (Agência Italiana de Medicamentos - Site: www.agenziafarmaco.gov.it/it/responsabili).
04.9 Overdose -
Uma dose única de 300 mcg / kg foi administrada por via subcutânea a um número limitado de voluntários saudáveis e em pacientes com câncer de pulmão não microcitoma, sem reações adversas graves. Os eventos adversos foram semelhantes aos ocorridos em indivíduos que receberam doses mais baixas de pegfilgrastim.
05.0 PROPRIEDADES FARMACOLÓGICAS -
05.1 "Propriedades farmacodinâmicas -
Grupo farmacoterapêutico: imunoestimulantes, fator estimulador de colônias.
Código ATC: L03AA13.
O fator estimulador de colônias de granulócitos humanos (G-CSF) é uma glicoproteína que regula a produção e liberação de neutrófilos da medula óssea. Pegfilgrastim é composto por uma molécula de G-CSF humano recombinante (r-metHuG-CSF) covalentemente ligada a uma única molécula de polietilenoglicol (PEG) de 20 kd. Pegfilgrastim é uma forma de filgrastim de longa duração devido à redução da depuração renal. O pegfilgrastim e o filgrastim têm mecanismos de ação idênticos, causando um aumento acentuado no número de neutrófilos periféricos em 24 horas, com aumentos desprezíveis nos monócitos e / ou linfócitos. Semelhante ao filgrastim, os neutrófilos produzidos em resposta ao pegfilgrastim exibem função normal ou aumentada, conforme demonstrado por avaliações da atividade quimiotática e fagocítica. Como outros fatores de crescimento hematopoiéticos, o G-CSF mostrou em vitro estimulando propriedades em células endoteliais humanas. G-CSF pode promover o crescimento em vitro de células mieloides, mesmo efeitos malignos e semelhantes podem ser detectados em vitro em algumas células não mieloides.
Em dois estudos randomizados, duplo-cegos e essenciais em pacientes com câncer de mama em estágio II-IV de alto risco submetidas a quimioterapia mielossupressora, incluindo doxorrubicina e docetaxel, o uso de pegfilgrastim como dose única uma vez por ciclo reduziu a duração da neutropenia e a incidência de neutropenia febril semelhante à observada com a administração diária de filgrastim (mediana de 11 dias de administração). Na ausência de suporte de fator de crescimento, este padrão foi relatado como resultando em uma duração média de neutropenia de grau 4 de 5-7 dias, com uma incidência de neutropenia febril de 30-40%. Em um estudo (n = 157) usando para uma dose fixa de 6 mg de pegfilgrastim, a duração média da neutropenia de grau 4 para o grupo do pegfilgrastim foi de 1,8 dias, em comparação com 1,6 dias no grupo do filgrastim (diferença de 0,23 dias, IC 95%: -0,15, 0,63). Durante todo o estudo. , a taxa de neutropenia febril foi de 13% dos pacientes tratados com pegfilgrastim em comparação com 20% dos pacientes tratados com filgrastim (diferença de 7%, IC de 95%: - 19%, 5%). Num segundo estudo (n = 310), utilizando uma dose ajustada ao peso (100 mcg / kg), a duração média da neutropenia de grau 4 no grupo do pegfilgrastim foi de 1,7 dias, em comparação com 1,8 dias no grupo do filgrastim. (Diferença 0,03 dias, IC 95%: -0,36, 0,30). A taxa geral de neutropenia febril foi de 9% dos pacientes tratados com pegfilgrastim e 18% dos pacientes tratados com filgrastim (diferença de 9%, IC de 95%: -16,8%, -1,1%).
Num estudo duplo-cego controlado por placebo em doentes com cancro da mama, o efeito do pegfilgrastim na incidência de neutropenia febril foi avaliado após a administração de um regime de quimioterapia associado a uma incidência de neutropenia febril de 10-20% (docetaxel 100 mg / m² a cada 3 semanas por 4 ciclos). Novecentos e vinte e oito pacientes foram randomizados para receber uma dose única de pegfilgrastim ou placebo aproximadamente 24 horas após a quimioterapia em cada ciclo (dia 2). A incidência de neutropenia febril foi menor em pacientes randomizados. a receber pegfilgrastim versus placebo (1% versus 17%, p
Um estudo de Fase II, randomizado, duplo-cego, de amostra limitada (n = 83) conduzido em pacientes submetidos à quimioterapia para leucemia mieloide aguda de novo comparou o pegfilgrastim (dose única de 6 mg) com o filgrastim, administrado durante a quimioterapia de indução. O tempo médio para a remissão da neutropenia grave foi de 22 dias em ambos os grupos de tratamento. O resultado a longo prazo não foi estudado (ver secção 4.4).
Em um estudo multicêntrico, randomizado e aberto de fase II (n = 37) em pacientes pediátricos com sarcoma, que receberam 100 mcg / kg de pegfilgrastim após o primeiro curso de quimioterapia com vincristina, doxorrubicina e ciclofosfamida (VAdriaC / IE), a maior duração de neutropenia grave (neutrófilos
05.2 "Propriedades farmacocinéticas -
A concentração sérica máxima de pegfilgrastim é observada 16 a 120 horas após a administração de uma dose subcutânea única; as concentrações séricas permanecem estáveis durante o período de neutropenia após quimioterapia mielossupressora. A eliminação do pegfilgrastim não é linear em relação à dose; a depuração sérica do pegfilgrastim diminui com o aumento da dose. O pegfilgrastim parece ser eliminado principalmente através da depuração mediada por neutrófilos, que é saturada em doses mais elevadas. De acordo com um mecanismo de depuração auto-regulado, a concentração sérica de pegfilgrastim diminui rapidamente em conjunto com o aumento dos neutrófilos.
Devido ao mecanismo de depuração mediado por neutrófilos, não é esperado que a insuficiência hepática ou renal afete a farmacocinética do pegfilgrastim. Num estudo aberto de dose única (n = 31), várias fases da insuficiência renal, incluindo a doença renal em fase terminal, não afetaram a farmacocinética do pegfilgrastim.
População idosa
Os dados limitados disponíveis indicam que a farmacocinética do pegfilgrastim em idosos (> 65 anos) é semelhante à dos adultos.
População pediátrica
A farmacocinética do pegfilgrastim foi estudada em 37 doentes pediátricos com sarcoma que receberam 100 μg / kg de pegfilgrastim após a conclusão da quimioterapia com VAdriaC / IE. O grupo de idade mais jovem (0-5 anos) teve maior "exposição média ao pegfilgrastim (AUC) (± desvio padrão) (47,9 ± 22,5 mcg • h / ml) do que crianças com mais de 6-11 anos e 12-21 anos (22,0 ± 13,1 mcg • h / ml e 29,3 ± 23,2 mcg • h / ml, respectivamente) (ver secção 5.1).
Com exceção do grupo de idade mais jovem (0-5 anos), a AUC média em pacientes pediátricos parecia semelhante à de pacientes adultos com câncer de mama de alto risco em estágio II-IV que receberam 100 mcg / kg de pegfilgrastim após a conclusão da doxorrubicina / docetaxel (ver seções 4.8 e 5.1).
05.3 Dados de segurança pré-clínica -
Os dados pré-clínicos de estudos tradicionais de toxicidade de dose repetida revelaram efeitos farmacológicos esperados, incluindo aumentos na contagem de glóbulos brancos, hiperplasia mieloide da medula óssea, hematopoiese extramedular e esplenomegalia.
Nenhum efeito adverso foi observado em ratos nascidos de mulheres grávidas aos quais pegfilgrastim foi administrado por via subcutânea, no entanto, em coelhos, pegfilgrastim administrado por via subcutânea causou toxicidade embriofetal (perda do embrião) em doses cumulativas de 4 vezes a dose recomendada para humanos. Estudos em ratos demonstraram que a passagem transplacentária do pegfilgrastim é possível. Estudos em ratos indicaram que a administração subcutânea de pegfilgrastim não teve efeito no desempenho reprodutivo, fertilidade, ciclo estral, dias entre o acasalamento e o coito e sobrevivência intra-uterina. A relevância destes dados para humanos é desconhecida.
06.0 INFORMAÇÕES FARMACÊUTICAS -
06.1 Excipientes -
Acetato de sódio *
Sorbitol (E420)
Polissorbato 20
Água para injetáveis
* O acetato de sódio é obtido por titulação de ácido acético glacial com hidróxido de sódio.
06.2 Incompatibilidade "-
Este medicamento não deve ser misturado com outros medicamentos, especialmente soluções de cloreto de sódio.
06.3 Período de validade "-
3 anos.
06.4 Precauções especiais de armazenamento -
Conservar no frigorífico (2 ° C - 8 ° C).
Neulasta pode ser conservado à temperatura ambiente (não superior a 30 ° C) uma vez e por um período máximo de 72 horas. Neulasta deixado à temperatura ambiente por mais de 72 horas deve ser descartado.
Não congele. A exposição acidental a temperaturas de congelamento, uma vez por menos de 24 horas, não afeta a estabilidade do Neulasta.
Manter o recipiente dentro da embalagem exterior para proteger o medicamento da luz.
06.5 Natureza da embalagem primária e conteúdo da embalagem -
Seringa pré-cheia (vidro Tipo I) com êmbolo de borracha e agulha de aço inoxidável com ou sem proteção automática da agulha.
A tampa da agulha da seringa pré-cheia contém borracha natural seca (um derivado do látex) (ver secção 4.4).
Cada seringa pré-cheia contém 0,6 ml de solução injetável. Embalagem de uma seringa pré-cheia, embalada com blister ou sem blister.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio -
Antes de administrar a solução de Neulasta, deve-se verificar a ausência de partículas visíveis, injetando-se apenas uma solução límpida e incolor.
Se agitado excessivamente, o pegfilgrastim pode formar agregados e tornar-se biologicamente inativo.
Deixe a seringa pré-cheia atingir a temperatura ambiente antes de injetar a solução.
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA "AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO" -
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
Holanda
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO -
EU / 1/02/227/001 1 embalagem de seringa com blister
035716012
EU / 1/02/227/002 embalagem de 1 seringa sem blister
EU / 1/02/227/004 1 embalagem de seringa com blister com proteção da agulha
035716036
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO -
Data da primeira autorização: 22 de agosto de 2002
Data da renovação mais recente: 16 de julho de 2007
10.0 DATA DE REVISÃO DO TEXTO -
Maio de 2015

















.jpg)