Ingredientes ativos: Ramipril
TRIATEC 1,25 mg comprimidos
Comprimidos TRIATEC 2,5 mg
TRIATEC 5 mg comprimidos
Comprimidos TRIATEC 10 mg
Indicações Por que o Triatec é usado? Para que serve?
TRITACE contém um medicamento denominado ramipril que pertence a um grupo de medicamentos denominados inibidores da ECA (inibidores da enzima de conversão da angiotensina).
TRIATEC atua:
- Ao diminuir a produção do corpo de substâncias que podem causar o aumento da pressão arterial
- Relaxando e dilatando seus vasos sanguíneos
- Tornando mais fácil para o coração bombear o sangue pelo corpo.
TRIATEC pode ser usado:
- Para tratar a hipertensão (hipertensão)
- Para reduzir o risco de um ataque cardíaco ou derrame
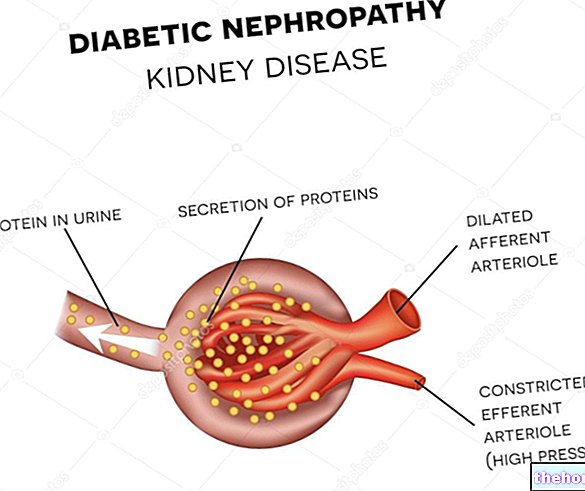
- Para reduzir o risco ou retardar o agravamento de problemas renais (seja você diabético ou não)
- Para tratar o coração quando ele não consegue bombear sangue suficiente para o resto do corpo (insuficiência cardíaca)
- Como tratamento após um ataque cardíaco (enfarte do miocárdio) quando associado a insuficiência cardíaca.
Contra-indicações Quando Triatec não deve ser usado
Não tome TRITACE:
- Se tem alergia (hipersensibilidade) ao ramipril, a outros medicamentos inibidores da ECA ou a qualquer outro componente do TRITACE listados na seção 6. Os sinais de uma reação alérgica podem ser erupção cutânea, dificuldade em engolir ou respirar, inchaço dos lábios, face, em a garganta ou língua
- Se alguma vez teve uma reação alérgica grave chamada 'angioedema'. Os sinais incluem coceira, erupção na pele (urticária), manchas vermelhas nas mãos, pés e garganta, inchaço da garganta e da língua, inchaço ao redor dos olhos e lábios, dificuldade em respirar e engolir
- Se está a fazer diálise ou a fazer algum outro tipo de filtração sanguínea. Dependendo do maquinário utilizado, o TRIATEC pode não ser adequado para você
- Se tem problemas renais devido ao fornecimento insuficiente de sangue aos rins (estenose da artéria renal).
- Durante os últimos 6 meses de gravidez (ver seção em "Gravidez e amamentação")
- Se a sua pressão arterial estiver excessivamente baixa ou instável. Seu médico precisará fazer esta avaliação
- Se tem diabetes ou insuficiência renal e está a ser tratado com um medicamento para baixar a tensão arterial contendo aliscireno.
Não tome TRITACE se alguma das condições acima se aplicar. Se não tiver certeza, pergunte ao seu médico antes de tomar TRITACE.
Precauções de uso O que você precisa saber antes de tomar Triatec
Verifique com seu médico ou farmacêutico antes de tomar TRITACE:
- Se você tem problemas de coração, fígado ou rins
- Se você perdeu muitos sais ou fluidos corporais (devido a indisposição, como vômitos, diarreia, sudorese excessiva ou por seguir uma dieta com baixo teor de sal, ou por tomar diuréticos por um longo período de tempo ou por ter sido submetido a uma diálise)
- Se você está prestes a se submeter a um tratamento para reduzir a alergia a picadas de abelha ou vespa (dessensibilização)
- Se estiver prestes a ser submetido a anestesia. Esta pode ser administrada para cirurgia ou tratamento dentário. Pode ser necessário interromper o tratamento com TRITACE no dia anterior; peça conselho ao seu médico
- Se você tem uma quantidade elevada de potássio no sangue (mostrado em uma análise de sangue)
- Está a tomar medicamentos ou encontra-se em condições tais que os seus níveis de sódio no sangue podem baixar. O seu médico pode pedir análises ao sangue em intervalos regulares, especialmente para verificar os seus níveis de sódio no sangue, especialmente se for idoso
- Se você tem uma doença do colágeno vascular, como esclerodermia ou lúpus eritematoso sistêmico.
- Deve informar o seu médico se pensa que está (ou pode vir a estar) grávida. TRIATEC não é recomendado nos primeiros 3 meses de gravidez e pode causar danos graves ao bebê após os 3 meses de gravidez (ver seção em "Gravidez e amamentação")
- Se estiver a tomar algum dos seguintes medicamentos para tratar a tensão arterial elevada: - um “antagonista dos recetores da angiotensina II” (ARAII) (também conhecido como sartans - por exemplo valsartan, telmisartan, irbesartan), particularmente se tiver problemas renais relacionados com diabetes - aliscireno "
O seu médico pode verificar a função renal, a pressão arterial e a quantidade de eletrólitos (como o potássio) no sangue em intervalos regulares. Consulte também as informações sob o título "Não tome TRITACE".
Crianças
A utilização de TRITACE não é recomendada em crianças e adolescentes com idade inferior a 18 anos porque a segurança e eficácia de TRITACE em crianças ainda não foram estabelecidas.
Se alguma das situações anteriores se aplicar a si (ou se não tiver a certeza), pergunte ao seu médico antes de tomar TRITACE.
Interações Quais medicamentos ou alimentos podem alterar o efeito de Triatec
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica (incluindo medicamentos à base de plantas). Isto porque TRITACE pode afetar o modo como alguns outros medicamentos atuam.
Além disso, alguns medicamentos podem afetar o modo como o TRITACE atua.
Informe o seu médico se estiver a tomar algum dos seguintes medicamentos. Estes medicamentos podem interferir com o TRITACE alterando sua ação:
- Medicamentos usados para aliviar a dor e a inflamação (por exemplo, medicamentos anti-inflamatórios não esteroidais (AINEs), como ibuprofeno, indometacina, aspirina)
- Medicamentos usados para tratar a pressão arterial baixa, choque, insuficiência cardíaca, asma ou alergias como a efedrina, noradrenalina ou adrenalina. Seu médico precisará verificar sua pressão arterial.
Informe o seu médico se estiver a tomar algum dos seguintes medicamentos. Estes medicamentos, quando tomados com TRITACE, podem aumentar a probabilidade de ocorrência de efeitos colaterais:
- Medicamentos usados para aliviar a dor e a inflamação (por exemplo, medicamentos anti-inflamatórios não esteroidais (AINEs), como ibuprofeno, indometacina, aspirina)
- Medicamentos para tratar o câncer (quimioterapia)
- Medicamentos para evitar a rejeição de órgãos após o transplante, como a ciclosporina
- Diuréticos como furosemida
- Medicamentos que podem aumentar a quantidade de potássio no sangue, como espironolactona, triamtereno, amilorida, sais de potássio e heparina (usados para tornar o sangue mais fluido)
- Medicamentos esteróides para o tratamento da inflamação, como prednisolona
- Alopurinol (usado para diminuir o teor de ácido úrico no sangue)
- Procainamida (para problemas de batimento cardíaco)
- Tome especial cuidado com TRIATEC ").
Informe o seu médico se estiver a tomar algum dos seguintes medicamentos. O mecanismo de ação desses medicamentos pode ser influenciado pelo TRITACE:
- Medicamentos para a diabetes, como hipoglicemiantes orais e insulina. TRITACE pode diminuir a quantidade de açúcar no sangue. Verifique cuidadosamente os seus níveis de açúcar no sangue quando tomar TRITACE.
- Lítio (para problemas psiquiátricos). TRITACE pode aumentar a quantidade de lítio no sangue. O nível de lítio no sangue deve ser verificado cuidadosamente pelo seu médico.
Se alguma das situações anteriores se aplicar a si (ou se não tiver a certeza), pergunte ao seu médico antes de tomar TRITACE.
Tomar TRITACE com alimentos e álcool
- Beber bebidas alcoólicas juntamente com TRITACE pode causar-lhe tonturas ou vertigens. Se quiser saber a quantidade de álcool a beber enquanto estiver a tomar TRITACE, converse com o seu médico, uma vez que os medicamentos usados para baixar a tensão arterial e o álcool podem ter. Receptor de angiotensina II (ARAII) ou aliscireno (ver também informação em "Não fazer tomar TRITACE "e" efeitos aditivos.
- TRIATEC pode ser tomado com ou fora das refeições.
Avisos É importante saber que:
Gravidez
Deve informar o seu médico se pensa que está (ou pode vir a estar) grávida.
Não deve tomar TRITACE nas primeiras 12 semanas de gravidez e não deve tomá-lo de todo após a 13ª semana, uma vez que o seu uso durante a gravidez pode ser prejudicial para o bebé. Se engravidar durante o tratamento com Triatec, informe o seu médico imediatamente.
Antes de planejar uma gravidez, deve-se mudar para outro medicamento mais adequado.
Hora da alimentação
Não deve tomar TRITACE se estiver a amamentar. Pergunte ao seu médico ou farmacêutico antes de tomar qualquer medicamento.
Condução e utilização de máquinas
Você pode sentir tonturas durante o tratamento com TRITACE. Isto é mais provável quando acabou de começar a tomar TRITACE ou acabou de aumentar a sua dose. Se isso acontecer, não conduza nem utilize quaisquer ferramentas ou máquinas.
Dose, Método e Tempo de Administração Como usar Triatec: Posologia
Tome TRITACE sempre de acordo com as indicações do médico. Deve consultar o seu médico ou farmacêutico se tiver dúvidas.
Tomando este remédio
- Tome este medicamento por via oral à mesma hora do dia todos os dias
- Engula os comprimidos inteiros com líquido.
- Não parta os comprimidos nem os mastigue.
Quanto voce tem que tomar
Tratamento da hipertensão
- A dose inicial usual é de 1,25 mg ou 2,5 mg uma vez ao dia.
- O seu médico ajustará a quantidade que você ingerir até que sua pressão arterial esteja sob controle.
- A dose máxima diária é de 10 mg.
- Se já está a tomar diuréticos, o seu médico pode interromper ou reduzir a quantidade antes de iniciar o tratamento com TRITACE.
Para reduzir o risco de um ataque cardíaco ou derrame
- A dose inicial é de 2,5 mg uma vez ao dia.
- O seu médico pode decidir aumentar a dosagem que você toma
- A dose usual é de 10 mg uma vez ao dia.
Tratamento para reduzir ou prevenir o agravamento dos problemas renais
- Você pode receber uma dose inicial de 1,25 mg ou 2,5 mg uma vez ao dia
- Seu médico ajustará a quantidade que você está tomando.
- A dose usual é de 5 mg ou 10 mg uma vez ao dia.
Tratamento de insuficiência cardíaca
- A dose inicial usual é de 1,25 mg uma vez ao dia.
- Seu médico ajustará a quantidade que você ingere.
- A dose máxima é de 10 mg por dia. É preferível dividir a dose em duas administrações diárias.
Tratamento após um ataque cardíaco
- A dose inicial usual é de 1,25 mg uma vez ao dia a 2,5 mg duas vezes ao dia.
- Seu médico ajustará a quantidade que você ingere.
- A dose usual é de 10 mg por dia. É preferível dividir a dose em duas administrações diárias.
Cidadãos idosos
O seu médico irá reduzir a dose inicial e ajustar o seu tratamento mais lentamente.
Overdose O que fazer se você tiver tomado muito Triatec
Se você tomar mais TRITACE do que deveria
Informe o seu médico ou vá ao pronto-socorro do hospital mais próximo. Não dirija até o hospital, peça a alguém para acompanhá-lo ou chame uma ambulância. Leve a caixa do medicamento com você. Isso porque seu médico precisa saber o que você contratou .
Se você se esquecer de tomar TRITACE
- Se você esquecer de uma dose, tome a sua dose normal quando for a hora.
- Não tome uma dose a dobrar para compensar um comprimido que se esqueceu.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
Efeitos colaterais Quais são os efeitos colaterais do Triatec
Como todos os medicamentos, TRITACE pode causar efeitos secundários, embora nem todas as pessoas os tenham.
Pare de tomar TRITACE e consulte o seu médico imediatamente se notar quaisquer efeitos secundários graves - pode necessitar de tratamento médico urgente:
- Edema da face, lábios ou garganta que torna difícil engolir ou respirar, bem como coceira ou erupção na pele. Isso pode ser um sinal de uma reação alérgica grave ao TRITACE.
- Reações cutâneas graves, incluindo erupção cutânea, úlceras bucais, agravamento de uma condição cutânea pré-existente, vermelhidão, bolhas e descamação da pele (como a síndrome de Stevens-Johnson, necrólise epidérmica tóxica ou eritema multiforme).
Informe o seu médico imediatamente se você tiver:
- Freqüência cardíaca mais rápida, batimento cardíaco irregular ou fortalecido (palpitações), dor no peito, aperto no peito ou problemas mais sérios, incluindo ataque cardíaco e derrame
- Falta de ar ou tosse. Estes podem ser sinais de problemas pulmonares
- Hematomas mais fáceis, sangramento mais longo do que o normal, quaisquer sinais de sangramento (por exemplo, sangramento nas gengivas), manchas roxas na pele ou início mais fácil de infecções, irritação na garganta e febre, sensação de cansaço, fraqueza, tontura ou pele pálida. Estes podem ser sinais de problemas de sangue ou medula óssea
- Dor de estômago forte que pode se estender para as costas. Isso pode ser um sinal de pancreatite (inflamação do pâncreas)
- Febre, arrepios, cansaço, perda de apetite, dor de estômago, enjoo, amarelecimento da pele ou dos olhos (icterícia). Estes podem ser sinais de problemas hepáticos, como hepatite (inflamação do fígado) ou lesões hepáticas.
Outros efeitos colaterais incluem:
Informe o seu médico se alguma das condições descritas abaixo se tornar grave ou persistir por mais do que alguns dias:
Comum (afetando menos de 1 paciente em cada 10 pacientes em terapia)
- Dor de cabeça ou sensação de cansaço
- Sentindo zonzo. É mais provável que isso aconteça quando a terapia com TRITACE acaba de começar ou a dose acaba de ser aumentada
- Fraqueza, hipotensão (pressão arterial anormalmente baixa), especialmente ao se levantar ou se levantar rapidamente
- Tosse seca irritante, inflamação dos seios da face (sinusite) ou bronquite, falta de ar
- Dor no estômago ou intestinos, diarreia, indigestão, mal-estar ou mal-estar
- Erupção cutânea com ou sem caroços
- Dores no peito
- Cãibras ou dores musculares
- Os exames de sangue mostram um nível de potássio mais alto do que o normal.
Incomum (afetando menos de 1 paciente em cada 100 pacientes em terapia)
- Problemas de equilíbrio (tonturas)
- Coceira e sensações incomuns na pele, como dormência, formigamento, queimação, ardência ou fricção (parestesia)
- Perda ou alteração do sabor
- Problemas de sono
- Humor deprimido, ansiedade, mais nervosismo do que o normal ou irritabilidade
- Nariz entupido, dificuldade em respirar ou piora da asma
- Edema do "intestino denominado" angioedema intestinal "e que se apresenta com sintomas como dor abdominal, vômitos e diarreia
- Azia, prisão de ventre ou boca seca
- Quantidade aumentada de urina durante o dia
- Mais suando do que o normal
- Perda ou diminuição do apetite (anorexia)
- Batimento cardíaco rápido ou irregular.
- Braços e pernas inchados. Isso pode ser um sinal de que seu corpo está segurando mais água do que o normal
- Rubor
- Visão embaçada
- Dor nas articulações
- Febre
- Impotência em homens, desejo sexual reduzido em homens e mulheres
- Um aumento no número de glóbulos brancos (eosinofilia) encontrados em exames de sangue
- Alterações na função do fígado, pâncreas ou rins demonstradas em análises ao sangue.
Raro (afetando menos de 1 paciente em cada 1000 pacientes em terapia)
- Sentindo-se fraco ou confuso
- Língua inchada e vermelha
- Descamação grave ou descamação da pele, erupção cutânea pustulosa com coceira
- Problemas nas unhas (como afrouxamento ou separação da unha de seu lugar)
- Erupção cutânea ou hematomas
- Manchas na pele e extremidades frias
- Olhos vermelhos, inchados ou lacrimejantes ou coceira
- Audição perturbada e zumbido no ouvido
- Sensação de fraqueza
- Diminuição do número de glóbulos vermelhos, brancos e plaquetas no sangue ou da concentração de hemoglobina, demonstrada em análises ao sangue.
Muito raro (afetando menos de 1 paciente em cada 10.000 pacientes em terapia)
- Maior consciência do sol.
Outros efeitos colaterais encontrados:
Informe o seu médico se alguma das condições descritas abaixo se tornar grave ou persistir por mais de alguns dias.
- Dificuldade de concentração
- Inchaço na boca
- Exames de sangue mostrando poucas células sanguíneas.
- Exames de sangue mostrando baixo teor de sódio no sangue
- Dedos e dedos que mudam de cor quando ficam frios e que formigam ou doem quando aquecidos (fenômeno de Raynaud)
- Aumento dos seios em homens
- Reações lentas ou alteradas
- Sensação de queimadura
- Mudança na percepção de odores
- Perda de cabelo
Se notar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico.
Expiração e retenção
Mantenha este medicamento fora do alcance e da vista das crianças. Não use Triatec após expirar o prazo de validade indicado nas embalagens, blisters e frascos. O prazo de validade refere-se ao último dia do mês indicado.
Este medicamento não requer quaisquer condições especiais de armazenamento.
Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico.
Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza.
Isso ajudará a proteger o meio ambiente.
Composição e forma farmacêutica
O que TRIATEC contém
O ingrediente ativo é ramipril
1,25 mg: Cada comprimido contém 1,25 mg de ramipril.
2,5 mg: Cada comprimido contém 2,5 mg de ramipril
5 mg: Cada comprimido contém 5 mg de ramipril
10 mg: Cada comprimido contém 10 mg de ramipril
Os outros ingredientes dos comprimidos são:
Comprimidos de 1,25 mg e 10 mg de hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearilfumarato de sódio.
2,5 mg comprimidos de hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearil fumarato de sódio, óxido de ferro amarelo E172.
Comprimidos de 5 mg hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearil fumarato de sódio, óxido de ferro vermelho E172.
Descrição da aparência do TRIATEC e conteúdo da embalagem
Comprimidos de 1,25 mg Comprimidos oblongos, brancos a esbranquiçados com linha de quebra, marcados na parte superior com 1,25 e logotipo da empresa e na parte inferior com HMN e 1,25. A pontuação serve apenas para facilitar a quebra, favorecendo a deglutição, e não para dividir o comprimido em doses iguais.
Comprimidos de 2,5 mg Comprimidos oblongos, amarelos a amarelados com linha de quebra, marcados na parte superior com 2,5 e o logotipo da empresa e na parte inferior com HMR e 2,5. O tablet pode ser dividido em partes iguais.
Comprimidos de 5 mg Comprimidos oblongos, vermelhos claros com ranhura, lado superior marcado com 5 e logotipo da empresa e lado inferior com HMP e 5. O comprimido pode ser dividido igualmente.
Comprimidos de 10 mg Comprimidos oblongos brancos a esbranquiçados com uma linha marcada na parte superior com HMO / HMO. O tablet pode ser dividido em partes iguais.
Triatec 1,25 mg comprimidos está disponível em embalagens de 14, 15, 20, 28, 30, 50, 90, 98, 100 comprimidos em blisters de PVC / alumínio e em embalagens de 500 comprimidos em frascos de vidro escuro com tampa.
Os comprimidos de Triatec 2,5 mg estão disponíveis em embalagens de 7, 10, 14, 15, 18, 20, 28, 30, 45, 50, 60, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio e em embalagens de 500 comprimidos em frascos de vidro escuro com tampa.
Os comprimidos de Triatec 5 mg estão disponíveis em embalagens de 10, 14, 15, 18, 20, 21, 28, 30, 45, 50, 56, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio e em embalagens de 500 comprimidos em frascos de vidro escuro com fecho.
Os comprimidos de Triatec 10 mg estão disponíveis em embalagens de 7, 10, 14, 15, 18, 20, 28, 30, 45, 50, 56, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio e em embalagens de 28, 56, 500 comprimidos em frascos de vidro escuro com tampa. Nem todos os tamanhos de embalagem podem ser comercializados
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
TABLETS TRIATEC
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Tablets
Cada comprimido contém 1,25 mg de ramipril.
Cada comprimido contém 2,5 mg de ramipril.
Cada comprimido contém 5 mg de ramipril.
Cada comprimido contém 10 mg de ramipril.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Comprimidos de 1,25 mg
Comprimidos oblongos brancos a esbranquiçados com ranhura, lado superior marcado com 1,25 e logotipo da empresa e lado inferior com HMN e 1,25. A pontuação serve apenas para facilitar a quebra, favorecendo a deglutição, e não para dividir o comprimido em doses iguais.
Comprimidos de 2,5 mg
Comprimidos oblongos amarelos a amarelados com vinco, lado superior com 2,5 e logotipo da empresa e lado inferior com HMR e 2,5. O tablet pode ser dividido em partes iguais.
Comprimidos de 5 mg
Comprimidos oblongos, vermelhos claros com linha vincada, lado superior com 5 e logotipo da empresa e lado inferior com HMP e 5. O comprimido pode ser dividido igualmente.
Comprimidos de 10 mg
Comprimidos oblongos brancos a esbranquiçados com ranhura, marcados na parte superior com HMO / HMO. O tablet pode ser dividido em partes iguais.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
- Tratamento da hipertensão.
- Prevenção cardiovascular: redução da morbidade e mortalidade cardiovascular em pacientes com:
• doenças cardiovasculares aterotrombóticas estabelecidas (doença cardíaca coronária anterior ou acidente vascular cerebral ou doença vascular periférica) ou
• diabetes com pelo menos um fator de risco cardiovascular (ver seção 5.1)
- Tratamento de doenças renais:
• Nefropatia glomerular diabética incipiente, definida pela presença de microalbuminúria
• Nefropatia glomerular diabética evidente, definida por macroproteinúria em pacientes com pelo menos um fator de risco cardiovascular (ver seção 5.1)
• Nefropatia glomerular não diabética evidente definida por macroproteinúria ≥3g / dia (ver secção 5.1).
- Tratamento da insuficiência cardíaca sintomática.
- Prevenção secundária após infarto agudo do miocárdio: redução da mortalidade após a fase aguda do infarto do miocárdio em pacientes com sinais clínicos de insuficiência cardíaca quando iniciada 48 horas após o início do infarto agudo do miocárdio
04.2 Posologia e método de administração
Uso oral.
Recomenda-se que TRITACE seja tomado à mesma hora todos os dias.
TRITACE pode ser tomado antes, durante ou após as refeições, porque a ingestão de alimentos não altera a sua biodisponibilidade (ver secção 5.2).
O TRIATEC deve ser engolido com um líquido e não deve ser mastigado ou esfarelado.
Adultos
Pacientes em tratamento com um diurético
Pode ocorrer hipotensão após o início do tratamento com TRITACE e é mais provável em pacientes tratados concomitantemente com um diurético. Portanto, recomenda-se precaução para esses pacientes, pois podem estar depletados no volume plasmático e / ou sais.
O diurético deve ser descontinuado, se possível, 2 ou 3 dias antes do início da terapêutica com TRITACE (ver secção 4.4).
Em pacientes hipertensos nos quais o diurético não foi descontinuado, a terapia com TRITACE deve ser iniciada com uma dose de 1,25 mg. A função renal e o potássio sérico devem ser monitorados. A dosagem subsequente de TRITACE deve ser ajustada de acordo com o valor de pressão arterial desejado.
Hipertensão
A dose deve ser individualizada de acordo com o perfil do paciente (ver seção 4.4) e controle da pressão arterial.
TRITACE pode ser usado sozinho ou em combinação com outras classes de medicamentos anti-hipertensivos (ver seções 4.3, 4.4, 4.5 e 5.1).
Dose inicial
O tratamento com TRITACE deve ser iniciado gradualmente, com uma dose inicial recomendada de 2,5 mg por dia.
Os doentes com hiperativação do sistema renina-angiotensina-aldosterona podem ter uma queda excessiva da pressão arterial após tomar a dose inicial. Para estes doentes, é recomendada uma dose inicial de 1,25 mg e iniciar o tratamento sob supervisão médica (ver secção 4.4) .
Titulação e dose de manutenção
A dose pode ser duplicada em intervalos de 2 a 4 semanas para atingir progressivamente o valor de pressão arterial necessário; a dose máxima de TRITACE é de 10 mg por dia. A dose geralmente é administrada uma vez ao dia.
Prevenção cardiovascular
Dose inicial
A dose inicial recomendada é 2,5 mg de TRITACE uma vez ao dia.
Titulação e dose de manutenção
A dosagem deve ser aumentada gradualmente no paciente com base na tolerabilidade do ingrediente ativo. Recomenda-se dobrar a dose após uma ou duas semanas de tratamento e - após mais duas a três semanas - aumentá-la até que a dose de manutenção alvo de 10 mg de TRITACE uma vez ao dia seja atingida.
Consulte também a posologia descrita acima para pacientes tratados com um diurético.
Tratamento de doenças renais
Em pacientes com diabetes e microalbuminúria:
Dose inicial
A dose inicial recomendada é de 1,25 mg de TRITACE uma vez ao dia.
Titulação e dose de manutenção
A dosagem deve ser aumentada gradualmente no paciente com base na tolerabilidade do ingrediente ativo.
Recomenda-se que a dose única diária seja duplicada para 2,5 mg após duas semanas e mais duas semanas para 5 mg.
Em pacientes com diabetes e pelo menos um fator de risco cardiovascular
Dose inicial
A dose inicial recomendada é 2,5 mg de TRITACE uma vez ao dia.
Titulação e dose de manutenção
A dosagem deve ser aumentada gradualmente no paciente com base na tolerabilidade do ingrediente ativo.
Recomenda-se que a dose única diária seja duplicada para TRITACE 5 mg após uma a duas semanas e, em seguida, para TRITACE 10 mg após mais duas a três semanas. A dose diária alvo é de 10 mg.
Em pacientes com nefropatia não diabética, definida por macroproteinúria ≥3g / dia
Dose inicial
A dose inicial recomendada é de 1,25 mg de TRITACE uma vez ao dia.
Titulação e dose de manutenção
A dosagem deve ser aumentada gradualmente no paciente com base na tolerabilidade do ingrediente ativo.
Recomenda-se que a dose única diária seja duplicada para 2,5 mg após duas semanas e, em seguida, para 5 mg após mais duas semanas.
Insuficiência cardíaca sintomática
Dose inicial
Em pacientes estabilizados com terapia diurética, a dose inicial recomendada é de 1,25 mg por dia.
Titulação e dose de manutenção
TRITACE deve ser titulado duplicando a dose a cada uma a duas semanas até uma dose diária máxima de 10 mg. Duas administrações por dia são preferíveis.
Prevenção secundária em pacientes com infarto agudo do miocárdio prévio e insuficiência cardíaca
Dose inicial
Após 48 horas de enfarte do miocárdio, em doentes clinicamente e hemodinamicamente estáveis, a dose inicial é 2,5 mg duas vezes ao dia durante três dias. Se a dose inicial de 2,5 mg não for tolerada, deve ser administrada uma dose. 1,25 mg duas vezes ao dia durante dois dias antes. aumentar para 2,5 mg e 5 mg duas vezes ao dia Se a dose não pode ser aumentada para 2,5 mg duas vezes ao dia, o tratamento deve ser interrompido.
Consulte também a posologia descrita acima para pacientes tratados com um diurético.
Titulação e dose de manutenção
A dose diária é subsequentemente aumentada duplicando-a em intervalos de um a três dias para uma dose de manutenção de 5 mg duas vezes ao dia.
Sempre que possível, a dose de manutenção é dividida em duas administrações por dia.
Se a dose não puder ser aumentada para 2,5 mg duas vezes ao dia, o tratamento deve ser interrompido. Ainda há experiência insuficiente no tratamento de pacientes com insuficiência cardíaca grave (NYHA IV) imediatamente após o infarto do miocárdio. Se for tomada a decisão de tratar esses pacientes, recomenda-se que a terapia seja iniciada com 1,25 mg uma vez ao dia e que se tenha cuidado especial em qualquer aumento da dose.
Populações especiais
Pacientes com insuficiência renal
A dose diária em pacientes com insuficiência renal deve ser baseada na depuração da creatinina (ver seção 5.2):
• se a depuração da creatinina for ≥ 60 ml / min, não é necessário ajustar a dose inicial (2,5 mg / dia); a dose máxima diária é de 10 mg;
• se a depuração da creatinina estiver entre 30-60 ml / min, não é necessário ajustar a dose inicial (2,5 mg / dia); a dose máxima diária é de 5 mg;
• se a depuração da creatinina estiver entre 10-30 ml / min, a dose inicial é 1,25 mg / dia e a dose diária máxima é 5 mg;
• Ramipril é pouco dialisável em pacientes hipertensos em hemodiálise; a dose inicial é de 1,25 mg / dia e a dose máxima diária é de 5 mg; o medicamento deve ser administrado algumas horas após a realização da diálise.
Doentes com insuficiência hepática (ver secção 5.2)
Em doentes com insuficiência hepática, o tratamento com TRITACE só deve ser iniciado sob supervisão médica rigorosa e a dose diária máxima de TRITACE é 2,5 mg.
Pacientes idosos
A dose inicial deve ser a mais baixa e a titulação subsequente deve ser muito gradual devido ao aumento da probabilidade de efeitos indesejáveis, particularmente em doentes muito idosos ou debilitados. Deve ser considerada uma dose inicial reduzida de ramipril de 1,25 mg.
População pediátrica
A segurança e eficácia do ramipril em crianças ainda não foram estabelecidas. Os dados atualmente disponíveis para Triatec são descritos nas seções 4.8, 5.1, 5.2 e 5.3, mas nenhuma recomendação específica sobre a posologia pode ser feita.
04.3 Contra-indicações
• Hipersensibilidade à substância ativa, a qualquer um dos excipientes ou a outros inibidores da ECA (inibidores da Enzima de Conversão da Angiotensina) (ver secção 6.1).
• História de angioedema (hereditário, idiopático ou angioedema prévio com inibidores da ECA ou ARAIIs).
• Tratamentos extracorpóreos que colocam o sangue em contato com superfícies carregadas negativamente (ver seção 4.5).
• Estenose significativa da artéria renal bilateral ou estenose unilateral em pacientes com um único rim funcionante.
• Segundo e terceiro trimestres de gravidez (ver secções 4.4 e 4.6).
• Ramipril não deve ser usado em pacientes com hipotensão ou hemodinamicamente instáveis
• A utilização concomitante de Triatec com medicamentos contendo aliscireno é contra-indicada em doentes com diabetes mellitus ou compromisso renal (TFG 2) (ver secções 4.5 e 5.1).
04.4 Advertências especiais e precauções adequadas de uso
Populações especiais
• Gravidez
A terapia com inibidores da ECA, como ramipril ou antagonistas dos receptores da angiotensina II (ARAIIs) não deve ser iniciada durante a gravidez.
Para pacientes que planejam gravidez, devem ser usados tratamentos anti-hipertensivos alternativos com um perfil de segurança comprovado para uso na gravidez, a menos que a terapia continuada com um inibidor da ECA / ARAIIs seja considerada essencial. Gravidez, o tratamento com inibidores da ECA / ARAIIs deve ser interrompido imediatamente e, se apropriado, deve ser iniciada uma terapêutica alternativa (ver secções 4.3 e 4.6).
• Pacientes particularmente em risco de hipotensão
- Pacientes com hiperativação do sistema renina-angiotensina-aldosterona
Pacientes com hiperativação do sistema renina-angiotensina-aldosterona correm o risco de uma queda aguda notável na pressão arterial e deterioração da função renal devido à inibição da ECA, especialmente quando o inibidor da ECA ou um diurético concomitante é administrado pela primeira vez. aumento da dose. A ativação relevante do sistema renina-angiotensina-aldosterona deve ser esperada e supervisão médica, incluindo monitoramento da pressão arterial, é necessária, por exemplo em:
• pacientes com hipertensão grave;
• pacientes com insuficiência cardíaca congestiva descompensada;
• pacientes com impedimento hemodinamicamente significativo para entrada ou saída do ventrículo esquerdo (por exemplo, estenose da válvula aórtica ou mitral);
• pacientes com estenose da artéria renal unilateral com um segundo rim funcional;
• pacientes nos quais existe ou pode ocorrer depleção de fluidos ou sais (incluindo pacientes em uso de diuréticos);
• pacientes com cirrose hepática e / ou ascite;
• durante uma grande cirurgia ou durante a anestesia com medicamentos que causam hipotensão.
Geralmente, é recomendado corrigir a desidratação, hipovolemia ou depleção de sal antes de iniciar o tratamento (no entanto, em pacientes com insuficiência cardíaca, esta ação corretiva deve ser cuidadosamente avaliada contra o risco de sobrecarga).
Bloqueio duplo do sistema renina-angiotensina-aldosterona (RAAS)
Existem evidências de que o uso concomitante de inibidores da ECA, bloqueadores do receptor da angiotensina II ou aliscireno aumenta o risco de hipotensão, hipercaliemia e diminuição da função renal (incluindo insuficiência renal aguda). O bloqueio duplo do SRAA através da utilização combinada de inibidores da ECA, bloqueadores dos receptores da angiotensina II ou aliscireno não é recomendado (ver secções 4.5 e 5.1).
Se a terapia de bloqueio duplo for considerada absolutamente necessária, isso só deve ser feito sob a supervisão de um especialista e com monitoramento próximo e frequente da função renal, eletrólitos e pressão arterial.
Os inibidores da ECA e os antagonistas dos receptores da angiotensina II não devem ser usados concomitantemente em pacientes com nefropatia diabética.
Insuficiência cardíaca transitória ou persistente após infarto do miocárdio
- Pacientes com risco de isquemia cardíaca ou cerebral em caso de hipotensão aguda
A fase inicial do tratamento requer supervisão médica cuidadosa.
• Pacientes idosos
Consulte a seção 4.2.
• Cirurgia
Se possível, recomenda-se que o tratamento com inibidores da enzima de conversão da angiotensina, como o ramipril, seja interrompido um dia antes da cirurgia.
• Monitoramento da função renal
A função renal deve ser avaliada antes e durante o tratamento e a dose deve ser ajustada principalmente nas primeiras semanas de tratamento. É necessária uma monitorização particularmente cuidadosa em doentes com compromisso renal (ver secção 4.2). C "é o risco de insuficiência renal, particularmente em pacientes com insuficiência cardíaca congestiva ou após transplante renal.
• Angioedema
Foram notificados casos de angioedema em doentes a receber inibidores da ECA, incluindo ramipril (ver secção 4.8).
Em caso de angioedema, TRITACE deve ser descontinuado.
O tratamento de emergência deve ser instituído imediatamente. Os pacientes devem ser mantidos em observação por pelo menos 12-24 horas e receber alta somente após a resolução completa dos sintomas.
Foi notificado angioedema intestinal em doentes a receber inibidores da ECA, incluindo TRITACE (ver secção 4.8). Esses pacientes apresentaram dor abdominal (com ou sem náuseas ou vômitos).
• Reações anafiláticas durante terapias de dessensibilização
A probabilidade e gravidade das reações anafiláticas ou anafilactóides após o contato com veneno de inseto ou outros alérgenos aumentam durante a terapia com inibidores da ECA. A descontinuação temporária de TRITACE deve ser considerada antes da dessensibilização.
• Monitoramento de eletrólitos: hipercalemia
Hipercalemia foi observada em alguns pacientes tratados com inibidores da ECA, incluindo TRITACE. Pacientes com risco de hipercalemia incluem aqueles com insuficiência renal, idade> 70 anos, diabetes mellitus não controlada ou aqueles que usam sais de potássio, diuréticos poupadores de potássio ou outras substâncias ativas que aumentam os níveis de potássio plasmático, ou condições como desidratação, insuficiência cardíaca aguda , acidose metabólica.
Se a utilização de qualquer uma das substâncias acima for considerada necessária, recomenda-se a monitorização regular do potássio sérico (ver secção 4.5).
• Monitoramento de eletrólitos: hiponatremia
Síndrome de secreção inapropriada de hormônio antidiurético (SIADH) e subsequente hiponatremia foi observada em alguns pacientes tratados com ramipril. Recomenda-se que os níveis séricos de sódio sejam monitorados regularmente em pacientes idosos e em outros pacientes com risco de hiponatremia.
• Neutropenia / agranulocitose
Neutropenia / agranulocitose, bem como trombocitopenia e anemia, foram raramente observadas, e também foi relatada depressão da medula óssea.
Recomenda-se monitorar o número de glóbulos brancos para permitir a detecção de uma possível leucopenia.
O monitoramento mais frequente é recomendado na fase inicial do tratamento e em pacientes com função renal comprometida, em pacientes com distúrbios do colágeno concomitantes (por exemplo, lúpus eritematoso ou esclerodermia) e naqueles tratados com medicamentos que podem causar alterações no hemograma (ver parágrafos 4.5 e 4.8).
• Diferenças étnicas
Os inibidores da ECA causam uma incidência maior de angioedema em pacientes negros do que em pacientes não negros.
Como outros inibidores da ECA, o ramipril pode ser menos eficaz na redução da pressão arterial em populações negras do que em populações não negras, possivelmente devido a uma maior prevalência de hipertensão com renina baixa em populações negras.
• Tosse
Foi relatada tosse com o uso de inibidores da ECA. Normalmente, a tosse é não produtiva, persistente e desaparece com a descontinuação da terapia. A tosse com inibidor da ECA deve ser considerada no diagnóstico diferencial da tosse.
04.5 Interações com outros medicamentos e outras formas de interação
Dados de ensaios clínicos mostraram que o bloqueio duplo do sistema renina-angiotensina-aldosterona (RAAS) por meio do uso combinado de inibidores da ECA, bloqueadores do receptor da angiotensina II ou aliscireno está associado a uma maior frequência de eventos adversos, como hipotensão, hipercaliemia e diminuição função renal (incluindo insuficiência renal aguda) em comparação com a utilização de um único agente ativo no sistema RAAS (ver secções 4.3, 4.4 e 5.1).
Associações contra-indicadas
Os tratamentos extracorpóreos que levam ao contato entre o sangue e superfícies carregadas negativamente, como diálise ou hemofiltração com membranas de alto fluxo (por exemplo, membranas de poliacrilonitrila) ou aférese de lipoproteína de baixa densidade por meio de sulfato de dextrano, são contra-indicados devido ao risco aumentado de reações anafilactoides graves (ver seção 4.3) Se este tipo de tratamento for necessário, o uso de diferentes membranas de diálise ou uma classe diferente de agentes anti-hipertensivos deve ser considerado.
Precauções para uso
Sais de potássio, heparina, diuréticos poupadores de potássio e outras substâncias ativas que aumentam os níveis de potássio no sangue (incluindo antagonistas da angiotensina II, trimetoprima, tacrolimus, ciclosporina):
Pode ocorrer hipercaliemia, portanto, é necessária uma monitoração cuidadosa dos níveis de potássio sérico.
Drogas anti-hipertensivas (por exemplo, diuréticos) e outras drogas com potencial efeito anti-hipertensivo (por exemplo, nitratos, antidepressivos tricíclicos, anestésicos, ingestão de álcool, baclofeno, alfuzosina, doxazosina, prazosina, tamsulosina, terazosina): Deve ser antecipada uma possível potenciação do risco de hipotensão (ver secção 4.2 para diuréticos).
Vasopressores simpaticomiméticos e outras substâncias (por exemplo, isoproterenol, dobutamida, dopamida, adrenalina) que podem reduzir o efeito anti-hipertensivo do TRIATEC: Recomenda-se o monitoramento da pressão arterial.
Alopurinol, imunossupressores, corticosteroides, procainamida, citostáticos e outros medicamentos que podem alterar o hemograma: aumento do risco de reações hematológicas (ver secção 4.4).
Sais de lítio: A excreção de lítio pode ser reduzida pelos inibidores da ECA e, portanto, a toxicidade do lítio pode ser aumentada.Os níveis séricos de lítio devem ser monitorados.
Agentes antidiabéticos, incluindo insulina: Podem ocorrer reações hipoglicêmicas. Portanto, o monitoramento da glicose no sangue é recomendado.
Antiinflamatórios não esteróides e ácido acetilsalicílico: deve ser antecipada uma possível redução do efeito anti-hipertensor de TRITACE Além disso, a terapêutica concomitante com inibidores da ECA e AINEs pode levar a um risco aumentado de agravamento da função renal e a um aumento da calemia.
04.6 Gravidez e amamentação
Gravidez
TRITACE não é recomendado durante o primeiro trimestre da gravidez (ver secção 4.4 e está contra-indicado durante o segundo e terceiro trimestres da gravidez (ver secção 4.3).
As evidências epidemiológicas sobre o risco de teratogenicidade após a exposição a inibidores da ECA durante o primeiro trimestre da gravidez não foram conclusivas; no entanto, um pequeno aumento no risco não pode ser excluído.
Para pacientes que planejam engravidar, devem ser usados tratamentos anti-hipertensivos alternativos com perfil de segurança comprovado para uso na gravidez, a menos que a terapia contínua com inibidores da ECA seja considerada essencial.
Quando a gravidez é diagnosticada, o tratamento com inibidores da ECA deve ser interrompido imediatamente e, se apropriado, uma terapia alternativa deve ser iniciada.
A exposição a inibidores da ECA / antagonistas do receptor da angiortensina II (ARAIIs) durante o segundo e terceiro trimestres em mulheres é conhecida por induzir toxicidade fetal (função renal diminuída, oligoidrâmnio, retardo de ossificação do crânio) e toxicidade neonatal (insuficiência renal, hipotensão, hipercaliemia) (ver secção 5.3 "Dados de segurança pré-clínica").
Caso a exposição ao inibidor da ECA tenha ocorrido a partir do segundo trimestre da gravidez, recomenda-se a verificação da função renal e do crânio por ultrassom.
Os recém-nascidos cujas mães tomaram inibidores da ECA devem ser cuidadosamente monitorizados quanto a hipotensão, oligúria e hipercaliemia (ver também secções 4.3 e 4.4).
Hora da alimentação
Dado que não existe informação suficiente sobre a utilização de ramipril durante a amamentação (ver secção 5.2), TRITACE não é recomendado e são preferíveis tratamentos alternativos com perfis de segurança mais bem estabelecidos durante a amamentação, especialmente quando amamenta um recém-nascido ou lactente.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
Alguns efeitos indesejáveis (por exemplo, sintomas de pressão arterial baixa, como tontura) podem interferir na capacidade do paciente de se concentrar e reagir e, portanto, representam um risco em situações em que essas habilidades são particularmente importantes (por exemplo, operar máquinas ou dirigir veículos).
Isso pode ocorrer principalmente no início do tratamento ou durante a substituição por outra terapia. Após a primeira dose ou aumento da dose, não é recomendado conduzir ou utilizar máquinas durante várias horas.
04.8 Efeitos indesejáveis
O perfil de segurança do ramipril inclui tosse seca persistente e reações devido à hipotensão.As reações adversas graves incluem angioedema, hipercaliemia, insuficiência hepática ou renal, pancreatite, reações cutâneas graves e neutropenia / agranulocitose.
A frequência dos efeitos indesejáveis é definida usando a seguinte convenção:
Muito comum (≥ 1/10); comum (≥ 1/100,
Dentro dos grupos de frequência, os efeitos indesejáveis são listados em ordem decrescente de gravidade.
População pediátrica
A segurança do ramipril foi monitorizada em 325 crianças e adolescentes, com idades entre 2 e 16 anos, em 2 estudos clínicos. Embora a natureza e a gravidade dos eventos adversos sejam semelhantes às dos adultos, a frequência dos seguintes eventos é maior em crianças:
Taquicardia, congestão nasal e rinite, "comum" (ou seja, ≥ 1/100,
Conjuntivite "comum" (ou seja, ≥ 1/100,
Tremor e urticária "incomum" (ou seja, ≥ 1/1000,
O perfil de segurança geral para ramipril em pacientes pediátricos não difere significativamente do perfil de segurança em adultos.
04.9 Overdose
Os sintomas associados à sobredosagem com inibidores da ECA podem incluir vasodilatação periférica excessiva (com hipotensão acentuada, choque), bradicardia, distúrbios eletrolíticos, insuficiência renal. Os doentes devem ser cuidadosamente monitorizados e o tratamento deve ser sintomático e de suporte. As principais medidas sugeridas incluem desintoxicação (lavagem gástrica, administração de adsorventes) e medidas para restaurar a estabilidade hemodinâmica, incluindo a administração de agonistas adrenais alfa 1 ou angiotensina II (angiotensinamida).O ramiprilato, o metabólito ativo do ramipril, é mal removido da circulação geral por hemodiálise.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: inibidores da ECA; Código A.T.C: C09AA05
Mecanismo de ação .
O ramiprilato, o metabólito ativo do pró-fármaco ramipril, inibe a enzima dipeptidilcarboxipeptidase I (sinônimos: enzima de conversão da angiotensina; quininase II). Esta enzima, em nível de plasma e tecido, determina a conversão da angiotensina I na substância vasoconstritora angiotensina II e a degradação do vasodilatador bradicinina.A formação reduzida de angiotensina II e a inibição da degradação da bradicinina levam à vasodilatação.
Uma vez que a angiotensina II também estimula a liberação de aldosterona, o ramiprilato causa uma redução na secreção de aldosterona.
A resposta média aos inibidores da ECA de pacientes hipertensos negros (afro-caribenhos) (geralmente essa população hipertensa tem um nível baixo de renina) é menor do que a de pacientes não negros.
Efeitos farmacodinâmicos .
Propriedades anti-hipertensivas:
A administração de ramipril causa uma redução acentuada na resistência arterial periférica. Geralmente, nem o fluxo plasmático renal nem a filtração glomerular sofrem alterações notáveis.
A administração de ramipril a pacientes hipertensos resulta em redução da pressão arterial tanto em pé quanto em supina, sem aumento compensatório da freqüência cardíaca.
Após uma dose oral única, na maioria dos pacientes a ação anti-hipertensiva ocorre após 1 ou 2 horas da ingestão, atinge seu efeito máximo após 3-6 horas e dura pelo menos 24 horas.
O efeito anti-hipertensivo máximo do tratamento contínuo com ramipril é geralmente alcançado após 3-4 semanas de tratamento.
Foi demonstrado que o efeito anti-hipertensivo é mantido por terapia prolongada por até 2 anos.
A descontinuação abrupta da terapia não causa um aumento rápido de rebote na pressão arterial.
Insuficiência cardíaca:
O ramipril tem se mostrado eficaz, além da terapia convencional com diuréticos e glicosídeos cardíacos, em pacientes com classes funcionais II-IV definidas pela New-York Heart Association. A droga teve efeitos benéficos na hemodinâmica cardíaca (diminuição da pressão de enchimento dos ventrículos esquerdo e direito, diminuição da resistência vascular periférica total, aumento do débito cardíaco e melhora do índice cardíaco). Também reduz a ativação neuroendócrina.
Eficácia clínica e segurança
Prevenção cardiovascular / nefroproteção:
Um estudo de prevenção controlado por placebo (o estudo HOPE) foi conduzido no qual ramipril foi adicionado à terapia padrão em mais de 9.200 pacientes. Pacientes com risco aumentado de doença cardiovascular resultante de doença cardiovascular aterotrombótica (doença arterial coronariana, acidente vascular cerebral ou doença vascular periférica) ou diabetes mellitus com pelo menos um fator de risco adicional (microalbuminúria documentada, hipertensão, alto nível de colesterol total, baixo nível de colesterol HDL, ou tabagismo), foram incluídos no estudo.
O estudo mostrou que o ramipril diminui de forma estatisticamente significativa a incidência de enfarte do miocárdio, morte cardiovascular e acidente vascular cerebral, isoladamente ou combinados (eventos primários combinados).
Estudo HOPE: principais resultados
O estudo MICRO - HOPE, um subestudo predefinido do estudo HOPE, avaliou o efeito da adição de 10 mg de ramipril ao regime atual versus placebo em 3.577 pacientes com idade ≥ 55 anos (sem limite superior de idade), a maioria com diabetes tipo 2 ( e pelo menos um outro fator de risco CV) normotenso ou hipertenso.
A análise primária dos resultados mostrou que 117 (6,5%) participantes tratados com ramipril e 149 (8,4%) tratados com placebo desenvolveram nefropatia evidente, o que corresponde a uma redução do risco relativo (RRR) de 24%. IC 95% [3 -40], p = 0,027.
O estudo REIN randomizado, duplo-cego, de grupo paralelo e controlado por placebo teve como objetivo demonstrar o efeito do tratamento com ramipril na taxa de redução da função glomerular (TFG) em 352 pacientes normotensos ou hipertensos (18-70 anos). idade) com proteinúria leve (ou seja, excreção urinária de proteínas> 1 e
A análise principal de pacientes com proteinúria mais grave (camada separada prematuramente devido ao benefício observado no grupo de ramipril) mostrou que a taxa média de diminuição da TFG por mês foi menor com ramipril do que com placebo; -0, 54 vs. - 0,88 mL / min / mês, p = 0,038. A diferença entre os grupos foi de 0,34 [0,03-0,65] por mês e aproximadamente 4 mL / min / ano; em 23, 1% dos pacientes no grupo de ramipril atingiu o desfecho secundário combinado de dobrar a concentração basal de creatinina sérica e / ou insuficiência renal em estágio final (ESRD) (necessidade de diálise ou transplante renal) versus 45,5% no placebo (p = 0,02).
Bloqueio duplo do sistema renina-angiotensina-aldosterona (RAAS):
Dois grandes ensaios clínicos randomizados (ONTARGET (ONgoing Telmisartan Alone e em combinação com Ramipril Global Endpoint Trial) e VA Nephron-D (The Veterans Affairs Nephropathy in Diabetes)) examinaram o uso da combinação de um inibidor da ECA com um antagonista do receptor de angiotensina II.
ONTARGET foi um estudo realizado em pacientes com história de doença cardiovascular ou cerebrovascular ou diabetes mellitus tipo 2 associada a evidência de lesão de órgãos. VA NEPHRON-D foi um estudo realizado em pacientes com diabetes mellitus tipo 2 e nefropatia diabética.
Esses estudos não demonstraram nenhum efeito benéfico significativo nos desfechos renais e / ou cardiovasculares e na mortalidade, enquanto um risco aumentado de hipercaliemia, lesão renal aguda e / ou hipotensão foi observado em comparação com a monoterapia.
Estes resultados também são relevantes para outros inibidores da ECA e antagonistas do receptor da angiotensina II, dadas as suas propriedades farmacodinâmicas semelhantes.
Os inibidores da ECA e os antagonistas dos receptores da angiotensina II não devem, portanto, ser usados simultaneamente em pacientes com nefropatia diabética.
ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) foi um estudo com o objetivo de verificar a vantagem de adicionar aliscireno à terapia padrão de um inibidor da ECA ou antagonista do receptor de angiotensina II em pacientes com diabetes mellitus. Tipo 2 e doença renal crônica , doença cardiovascular ou ambos. O estudo foi encerrado precocemente devido a um risco aumentado de eventos adversos. A morte cardiovascular e o acidente vascular cerebral foram numericamente mais frequentes no grupo de aliscireno do que no grupo de placebo, e eventos adversos e eventos adversos graves de interesse ( hipercaliemia, hipotensão e disfunção renal) foram relatados com mais freqüência no grupo aliscireno do que no grupo placebo.
Prevenção secundária após infarto agudo do miocárdio
O estudo AIRE incluiu mais de 2.000 pacientes com sinais clínicos transitórios / persistentes de insuficiência cardíaca após infarto do miocárdio documentado. O tratamento com ramipril começou 3-10 dias após o infarto agudo do miocárdio. O estudo indicou que, após um tempo médio de acompanhamento de 15 meses, a mortalidade em pacientes tratados com ramipril foi de 16,9%, enquanto em pacientes tratados com ramipril. Tratados com placebo foi de 22,6%, o que significa uma redução absoluta na mortalidade de 5,7% e uma redução do risco relativo de 27% (IC de 95% [11-40%]).
População pediátrica
Em um ensaio clínico randomizado, duplo-cego, controlado por placebo envolvendo 244 pacientes pediátricos com hipertensão (73% hipertensão primária), com idades entre 6 e 16 anos, os pacientes receberam doses baixas baseadas no peso, níveis médios ou altos de ramipril para atingir o ramiprilato plasmático concentrações correspondentes às dosagens para adultos de 1,25 mg, 5 mg e 20 mg. Ao final de 4 semanas, o ramipril foi ineficaz na redução da pressão arterial sistólica, mas na dose mais elevada ele reduziu a pressão arterial diastólica. Tanto as doses médias quanto as altas de ramipril mostraram uma redução significativa na pressão arterial sistólica e diastólica em crianças com hipertensão confirmada.
Este efeito não foi observado em um estudo clínico randomizado, duplo-cego, de escalonamento de dose interrompido após 4 semanas em 218 pacientes pediátricos de 6 a 16 anos (75% de hipertensão primária), onde as pressões sistólica e diastólica demonstraram rebote modesto, mas não retorno estatisticamente significativo à linha de base, em todos os três níveis de dosagem de ramipril testados para peso [dose baixa (0,625 mg - 2,5 mg), dose média (2,5 mg) mg - 10 mg) ou dose alta (5 mg - 20 mg)]. O ramipril não mostrou uma relação linear dose / resposta na população pediátrica estudada.
05.2 Propriedades farmacocinéticas
Farmacocinética e Metabolismo
Absorção
Após a administração oral, o ramipril é rapidamente absorvido pelo trato gastrointestinal: a concentração plasmática máxima de ramipril é atingida em 1 hora. Com base na recuperação urinária, a absorção é de pelo menos 56% e não é significativamente afetada pela presença de alimentos no trato gastrointestinal. A biodisponibilidade do metabolito ativo ramiprilato após administração oral de 2,5 mg e 5 mg de ramipril é de 45%.
As concentrações plasmáticas máximas de ramiprilato, o único metabólito ativo de ramipril, são atingidas 2-4 horas após a ingestão de ramipril. As concentrações plasmáticas de ramiprilato no estado estacionário após a administração uma vez ao dia das doses diárias usuais de ramipril são atingidas no quarto dia de tratamento aproximadamente .
Distribuição
A ligação do ramipril às proteínas séricas é de aproximadamente 73% e do ramiprilato de aproximadamente 56%.
Metabolismo
O ramipril é quase completamente metabolizado em ramiprilato e o éster de dicetopiperazina, a forma ácida da dicetopiperazina e os glicuronídeos de ramipril e ramiprilato.
Eliminação
A excreção dos metabolitos ocorre principalmente por via renal.
As concentrações plasmáticas de ramiprilato diminuem de maneira polifásica. Devido à sua ligação potente e saturável à ECA e à dissociação lenta da enzima, o ramiprilato exibe uma fase de eliminação terminal prolongada em concentrações plasmáticas muito baixas.
Após múltiplas doses diárias de ramipril, a meia-vida efetiva das concentrações de ramiprilato foi de 13-17 horas para as doses de 5-10 mg e maior para as doses inferiores de 1,25-2,5 mg. Essa diferença está relacionada à capacidade de saturação da enzima para vincular ramiprilato.
Uma única dose oral de ramipril produziu um nível indetectável de ramipril e seu metabólito no leite materno. No entanto, o efeito da administração de doses múltiplas não é conhecido.
Doentes com insuficiência renal (ver secção 4.2)
A excreção renal do ramiprilato é reduzida em pacientes com insuficiência renal e a depuração renal do ramiprilato é proporcional à depuração da creatinina, resultando em concentrações plasmáticas elevadas de ramiprilato que diminuem mais lentamente do que em pacientes com função renal normal.
Doentes com insuficiência hepática (ver secção 4.2)
Em pacientes com função hepática comprometida, a metabolização de ramipril em ramiprilato é retardada devido à diminuição da atividade das esterases hepáticas; nesses pacientes, os níveis plasmáticos de ramipril estão aumentados. As concentrações máximas de ramiprilato nesses pacientes, no entanto, não são diferentes daquelas observada em indivíduos com função hepática normal.
Hora da alimentação
Uma única dose oral de ramipril produziu um nível indetectável de ramipril e seu metabólito no leite materno. No entanto, o efeito da administração de doses múltiplas não é conhecido.
População pediátrica
O perfil farmacocinético do ramipril foi estudado em 30 doentes pediátricos hipertensos, com idades entre 2 a 16 anos e peso ≥ 10 kg. Após a administração de doses de 0,05 a 0,2 mg / kg, o ramipril foi rápida e amplamente metabolizado em ramiprilato. As concentrações plasmáticas máximas de ramiprilato ocorreram em 2-3 horas. A depuração de ramiprilato está altamente correlacionada com o log do peso corporal (p
05.3 Dados de segurança pré-clínica
A administração oral de ramipril foi considerada isenta de toxicidade aguda em roedores e cães. Estudos envolvendo administração oral crônica foram conduzidos em ratos, cães e macacos. Alterações de eletrólitos plasmáticos foram detectadas nas três espécies. Como uma expressão da atividade farmacodinâmica do ramipril, um aumento pronunciado do aparelho justaglomerular foi encontrado em cães e macacos, começando com doses diárias de 250 mg / kg. Ratos, cães e macacos toleraram doses diárias de 2, 2,5 e 8 mg / kg, respectivamente, sem efeitos adversos.
Danos renais irreversíveis foram observados em ratos muito jovens tratados com uma única dose de ramipril.
Os estudos de toxicologia reprodutiva em ratos, coelhos e macacos não revelaram propriedades teratogénicas. A fertilidade não foi afetada em ratos machos ou fêmeas.
A administração de ramipril a ratas durante o período de gestação e lactação resultou em dano renal irreversível (dilatação da pelve renal) na prole em doses diárias de 50 mg / kg de peso corporal ou superior.
O teste de mutagenicidade, conduzido usando vários sistemas de teste, não forneceu evidências de que o ramipril possui propriedades mutagênicas ou genotóxicas.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Comprimidos de 1,25 mg
hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearilfumarato de sódio.
Comprimidos de 2,5 mg
hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearilfumarato de sódio, óxido de ferro amarelo E172.
Comprimidos de 5 mg
hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearil fumarato de sódio, óxido de ferro vermelho E 172.
Comprimidos de 10 mg
hipromelose, amido de milho pré-gelatinizado, celulose microcristalina, estearilfumarato de sódio.
06.2 Incompatibilidade
Não é relevante.
06.3 Período de validade
3 anos.
06.4 Precauções especiais para armazenamento
Este medicamento não requer quaisquer condições especiais de armazenamento
06.5 Natureza da embalagem primária e conteúdo da embalagem
1,25 mg: embalagens de 14, 15, 20, 28, 30, 50, 90, 98, 100 comprimidos em blisters de PVC / alumínio
2,5 mg: embalagens de 7, 10, 14, 15, 18, 20, 28, 30, 45, 50, 60, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio,
5 mg: embalagens de 10, 14, 15, 18, 20, 21, 28, 30, 45, 50, 56, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio
10 mg: embalagens de 7, 10, 14, 15, 18, 20, 28, 30, 45, 50, 56, 90, 98, 99, 100, 300, 320, 500 comprimidos em blisters de PVC / alumínio
1,25 mg: 500 comprimidos em frasco de vidro escuro tipo III (Eur.Ph.) com tampa de rosca de HDPE.
2,5 mg: 500 comprimidos em frasco de vidro escuro tipo III (Eur.Ph.) com tampa de rosca de HDPE.
5 mg: 500 comprimidos em frasco de vidro escuro tipo III (Eur.Ph.) com tampa de rosca de HDPE.
10 mg: 25, 56, 500 comprimidos em frasco de vidro escuro tipo III (Eur.Ph.) com tampa de rosca de HDPE.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Sanofi S.p.A. - Viale L. Bodio, 37 / B - Milão 20158
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
"Comprimidos de 1,25 mg" - 28 comprimidos em PVC / AL A.I.C. n.: 027161049
"Comprimidos de 2,5 mg" - 28 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161052
"Comprimidos de 2,5 mg" - 320 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161088
"Comprimidos de 5 mg" - 14 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161064
"Comprimidos de 5 mg" - 320 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161090 "comprimidos de 10 mg" - 28 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161076
"Comprimidos de 10 mg" - 320 comprimidos divisíveis em PVC / AL A.I.C. n.: 027161102
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 1 de março de 1990 Triatec 1,25 mg, 2,5 mg, 5 mg.
23 de fevereiro de 2004 Triatec 10 mg
Última data de renovação: 01 de junho de 2010
10.0 DATA DE REVISÃO DO TEXTO
Maio de 2015