Generalidade
O termo retinite pigmentosa (RP) identifica um grupo de doenças genéticas caracterizadas por degeneração retiniana progressiva.

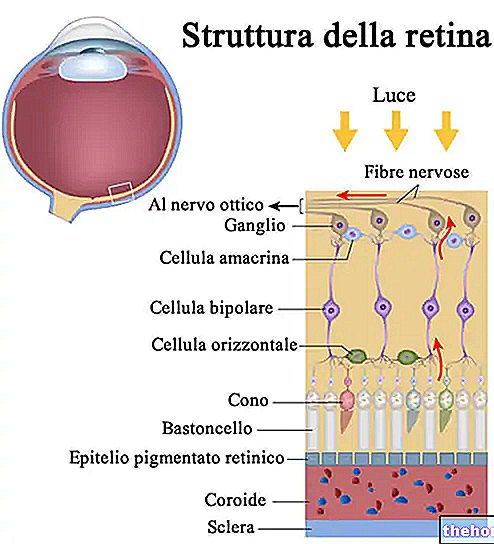
A retinite pigmentosa é uma distrofia retiniana caracterizada pela perda gradual de fotorreceptores e disfunção do epitélio pigmentar, o que significa que a retina reduz progressivamente sua capacidade de transmitir informações visuais ao cérebro através do nervo óptico.
O processo patológico começa com alterações do epitélio pigmentar da retina. À medida que a retinite pigmentosa progride, ocorre um adelgaçamento dos vasos sanguíneos que irrigam a retina, que sofrem atrofia. Ao examinar o fundo, os depósitos característicos são detectáveis visualmente. Pigmento retinal ( daí o nome da doença). Alterações atróficas e danos também podem envolver o nervo óptico e, gradualmente, as células fotossensíveis da retina morrem.
Pacientes afetados por retinite pigmentosa inicialmente experimentam problemas de visão, especialmente em ambientes mal iluminados e se queixam de uma constrição do campo visual periférico. A visão central é poupada até os estágios finais da doença, e o resultado final pode variar dramaticamente: muitas pessoas com retinite pigmentosa mantêm a visão limitada ao longo da vida, enquanto outras perdem totalmente a visão.
A retinite pigmentosa é uma doença hereditária, causada principalmente por alterações genéticas transmitidas por um ou ambos os pais. O tipo de defeito genético determina quais células da retina estão mais envolvidas no distúrbio e permite distinguir, do ponto de vista clínico, as diferentes condições. Até o momento, mais de 50 defeitos genéticos diferentes implicados na retinite pigmentosa foram identificados. As anormalidades podem ser transmitidas de pais para filhos por meio de um dos três padrões de herança: autossômica recessiva, autossômica dominante ou heterossômica recessiva (ligada ao X ou ligada ao X).
Sintomas
Para mais informações: Sintomas de retinite pigmentosa
A retinite pigmentosa é geralmente encontrada em adolescentes e adultos jovens. Os sintomas costumam aparecer entre as idades de 10 e 30 anos, mas o diagnóstico pode ser feito na primeira infância ou muito mais tarde na vida.
Os primeiros sintomas de retinite pigmentosa podem incluir:
- Dificuldade em enxergar à noite (cegueira noturna) ou em condições de pouca luz
- Adaptação lenta da visão no escuro para a luz e vice-versa;
- Estreitamento do campo visual e perda da visão periférica;
- Sensibilidade à luz e ao brilho.
Alguns sintomas dependem do tipo de fotorreceptores envolvidos. Os bastonetes são responsáveis pela visão em preto e branco, enquanto os cones permitem distinguir as cores.
Na maioria dos casos de retinite pigmentosa, os bastonetes são envolvidos primeiro. No entanto, nas formas de evolução rápida, os cones também podem ser afetados em um estágio inicial.
Os bastonetes estão concentrados nas partes externas da retina e são ativados pela luz fraca, de modo que sua degeneração afeta a visão periférica e noturna. Se os cones estiverem envolvidos, é possível experimentar a perda da percepção das cores e da visão central.
A predominância de fotorreceptores envolvidos é determinada pelo defeito particular presente na composição genética do paciente.
Freqüentemente, o primeiro sintoma da retinite pigmentosa é a cegueira noturna (ou noctalopia). Algumas pessoas acham que precisam de cada vez mais tempo para se ajustar às diferenças de luz à medida que passam de uma área bem iluminada para uma mais escura. Uma forma típica de perda de visão induz o estreitamento da visão periférica (visão em túnel ou telescópio); este padrão é denominado escotoma em anel. Às vezes, esse fenômeno pode estar ausente nos estágios iniciais, mas é percebido quando o indivíduo frequentemente tropeça em objetos ou se envolve em um acidente de trânsito. Quando a perda de visão envolve a área central da retina (também chamada de distrofia macular) dos pacientes têm dificuldade para ler e trabalhos detalhados que exigem concentração em um único objeto, como enfiar um fio no buraco de uma agulha. Muitos pacientes relatam ter visto flashes de luz (fotopsia), muitas vezes descritos como luzes pequenas e cintilantes.
A taxa de progressão da doença e o grau de perda visual variam de pessoa para pessoa. Alguns casos extremos podem evoluir rapidamente em duas décadas, outros um curso lento que nunca leva à cegueira completa. O início precoce é encontrado em formas mais graves de retinite pigmentosa, enquanto os pacientes com condições mais brandas (por exemplo, autossômica dominante) podem desenvolver a doença na quinta ou sexta década de vida. Em famílias com retinite pigmentar ligada ao X, os homens são afetados com mais frequência do que as mulheres e mais gravemente; as mulheres, por outro lado, transmitem a característica genética (carregam o gene alterado no cromossomo X) e manifestam os sintomas da doença com menos frequência.
Complicações
A retinite pigmentosa continuará a progredir, embora lentamente. No entanto, a cegueira completa é rara, mas pode ocorrer redução significativa da visão periférica e central.
Pacientes com retinite pigmentosa freqüentemente desenvolvem inchaço da retina (edema macular) ou catarata em idade precoce. Essas complicações podem ser tratadas se interferirem na visão.
Doenças relacionadas
Comumente, um paciente com retinite pigmentosa não apresenta outros distúrbios e, neste caso, falamos de retinite pigmentosa "não sindrômica" ou simples. No entanto, várias síndromes compartilham alguns sintomas clínicos com esta doença ocular; a mais comum é a síndrome de Usher, que afeta aproximadamente 10-30% de todos os pacientes com retinite pigmentosa e está associada à perda auditiva congênita ou progressiva concomitante. Na amaurose congênita de Leber, no entanto, as crianças podem ficar cegas ou quase cegas nos primeiros seis meses de vida. Outras doenças relacionadas à retinite pigmentosa incluem a síndrome de Bardet-Biedl e a doença de Refsum.
Causas
A doença pode ser causada por uma série de defeitos genéticos: na verdade, existem vários genes que, se afetados pela alteração, podem causar o fenótipo da retinite pigmentosa. Normalmente codificam proteínas envolvidas na cascata de transdução que permite a visão, fatores a transcrição celular (que enviam mensagens errôneas para as células da retina) ou para os elementos que compõem a estrutura dos fotorreceptores. Mutações genéticas herdadas estão presentes nas células desde o momento da concepção; anormalidades comuns incluem aquelas dos genes RP1 (na retinite pigmentosa-1, autossômica dominante) , RHO (RP4, autossômica dominante) e RDS (RP7, autossômica dominante). Causas não hereditárias de retinite pigmentosa são raras, mas a possibilidade de encontrar um caso isolado (mutação espontânea), em que não há história familiar de a doença.