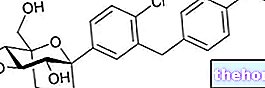
Ingredientes ativos: Prucaloprida
Comprimidos revestidos por película Resolor 1 mg
Comprimidos revestidos por película Resolor 2 mg
Indicações Por que o Resolor é usado? Para que serve?
O Resolor contém o ingrediente ativo prucaloprida.
O Resolor pertence a um grupo de medicamentos que melhoram a motilidade intestinal (procinéticos gastrointestinais). Atua na parede muscular do intestino, ajudando a restaurar seu funcionamento normal. O Resolor é usado no tratamento da constipação crônica em adultos nos quais os laxantes não atuam corretamente.
Não use em crianças e adolescentes com menos de 18 anos.
Contra-indicações Quando Resolor não deve ser usado
Não tome Resolor
- se você é alérgico à prucaloprida ou a qualquer outro componente deste medicamento (listados na seção 6),
- se você está passando por diálise renal,
- se tiver uma perfuração ou obstrução da parede intestinal, inflamação grave do trato intestinal, como doença de Crohn, colite ulcerosa ou megacólon / megareto tóxico.
Precauções de uso O que você precisa saber antes de tomar o Resolor
Fale com o seu médico antes de tomar o Resolor.
Tome especial cuidado com o Resolor e diga ao seu médico:
- se você tem doença renal grave,
- se você tem doença hepática grave,
- se você está atualmente sob supervisão médica para um problema sério de saúde, como uma doença cardíaca ou pulmonar, um distúrbio mental ou do sistema nervoso, câncer, AIDS ou um distúrbio hormonal.
Interações Quais medicamentos ou alimentos podem modificar o efeito do Resolor
Informe o seu médico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos.
Resolor com comida e bebida
O Resolor pode ser tomado com ou sem alimentos e bebidas a qualquer hora do dia.
Avisos É importante saber que:
Gravidez e amamentação
Resolor não é recomendado durante a gravidez.
- Informe o seu médico se estiver grávida ou planejando engravidar.
- Use um método contraceptivo confiável durante o tratamento com Resolor para evitar engravidar.
- Se engravidar durante o tratamento com Resolor, informe o seu médico.
Durante a amamentação, a prucaloprida pode passar para o leite materno. A amamentação não é recomendada durante o tratamento com Resolor. Consulte seu médico sobre isso.
Peça conselho ao seu médico antes de tomar qualquer medicamento.
Condução e utilização de máquinas
É improvável que o Resolor afete a sua capacidade de conduzir ou utilizar máquinas. No entanto, em alguns casos, o Resolor pode causar tonturas e cansaço, principalmente no primeiro dia de tratamento, o que pode afetar a condução e utilização de máquinas.
Resolor contém lactose
Se foi informado pelo seu médico que tem "intolerância a alguns açúcares, contacte-o antes de tomar este medicamento.
Dose, método e tempo de administração. Como usar o Resolor: Posologia
Tome sempre este medicamento exatamente como descrito neste folheto ou conforme indicado pelo seu médico.
Em caso de dúvida, consulte o seu médico ou farmacêutico. Continue a tomar Resolor todos os dias durante o tempo definido pelo seu médico. O seu médico pode querer reavaliar a sua condição e os benefícios do tratamento prolongado após as primeiras 4 semanas e a intervalos regulares daí em diante.
A dose usual de Resolor para a maioria dos pacientes é um comprimido de 2 mg uma vez ao dia.
Se você tem mais de 65 anos ou tem doença hepática grave, a dose inicial é um comprimido de 1 mg uma vez ao dia; se necessário, o seu médico pode aumentar a dose para 2 mg uma vez por dia.
O seu médico também pode recomendar a dose mais baixa de um comprimido de 1 mg uma vez por dia se tiver doença renal grave.
Tomar mais do que a dose recomendada não aumentará a eficácia do medicamento.
O Resolor é indicado apenas para adultos e não deve ser tomado por crianças ou adolescentes até aos 18 anos de idade.
Overdose O que fazer se você tiver tomado muito Resolor
Se você tomar mais Resolor do que deveria
É importante respeitar a dose prescrita pelo seu médico. Se você tomou mais Resolor do que o prescrito, pode sentir diarreia, dor de cabeça e / ou náuseas. Em caso de diarreia, beba bastante água.
Se você esquecer de tomar o Resolor
Não tome uma dose a dobrar para compensar um comprimido que se esqueceu. Tome a sua próxima dose à hora habitual.
Se você parar de usar o Resolor
Se você parar o Resolor, os sintomas de constipação podem retornar.
Caso ainda tenha dúvidas sobre o uso deste medicamento, fale com o seu médico ou farmacêutico
Efeitos colaterais Quais são os efeitos colaterais do Resolor
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham. Os efeitos indesejáveis ocorrem principalmente no início do tratamento e geralmente desaparecem alguns dias após a continuação do tratamento.
Os seguintes efeitos secundários ocorreram com muita frequência (podem afetar mais de 1 em 10 pessoas): dores de cabeça, náuseas, diarreia e dor abdominal.
Os seguintes efeitos secundários ocorreram com frequência (podem afetar até 1 em 10 pessoas): diminuição do apetite, tonturas, vômitos, distúrbios digestivos (dispepsia), flatulência, borborigmas intestinais anormais, cansaço.
Os seguintes efeitos secundários pouco frequentes (podem afetar até 1 em 100 pessoas) também foram notificados: tremores, palpitações, hemorragia rectal, aumento da frequência de micção (polaciúria), febre e enjoo. Se ocorrer palpitações, informe o seu médico.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico, incluindo quaisquer efeitos secundários possíveis não mencionados neste folheto. Você também pode relatar os efeitos colaterais diretamente através do sistema nacional de notificação listado no Apêndice V. Ao relatar os efeitos colaterais, você pode ajudar a fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no blister e na embalagem exterior após VAL. O prazo de validade corresponde ao último dia desse mês.
Conservar na embalagem original para proteger da humidade.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
O que Resolor contém
O ingrediente ativo é a prucaloprida.
Um comprimido revestido por película de Resolor 1 mg contém 1 mg de prucaloprida (como succinato).
Um comprimido revestido por película de Resolor 2 mg contém 2 mg de prucaloprida (como succinato).
Os outros ingredientes são:
Lactose monohidratada (ver seção 2), celulose microcristalina, dióxido de sílica coloidal, estearato de magnésio, hipromelose, triacetina, dióxido de titânio (E171), macrogol. O comprimido de 2 mg também contém óxido de ferro vermelho (E172), óxido de ferro amarelo (E172), laca de alumínio índigo carmim (E132).
Descrição da aparência do Resolor e conteúdo da embalagem
Os comprimidos revestidos por película de Resolor 1 mg são brancos a esbranquiçados, redondos, marcados com “PRU 1” num dos lados.
Os comprimidos revestidos por película de Resolor 2 mg são comprimidos redondos, cor-de-rosa, com a gravação “PRU 2” numa das faces.
O Resolor está disponível em blister de alumínio / alumínio perfurado para dose unitária (calendário) contendo 7 comprimidos. Cada embalagem contém 7x1, 14x1, 28x1 ou 84x1 comprimidos revestidos por película.
Nem todos os tamanhos de embalagem podem ser comercializados.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
RESOLOR 1 MG COMPRIMIDOS REVESTIDOS COM PELÍCULA
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Cada comprimido revestido por película contém 1 mg de prucaloprida (como succinato).
Excipientes com efeito conhecido: Cada comprimido revestido por película contém 142,5 mg de lactose (na forma mono-hidratada).
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Comprimido revestido por película (comprimido).
Comprimidos brancos a esbranquiçados, redondos, biconvexos, com a gravação “PRU 1” numa das faces.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
O Resolor é indicado para o tratamento sintomático da constipação crônica em adultos para os quais os laxantes não proporcionam alívio adequado.
04.2 Posologia e método de administração
Dosagem
Adultos: 2 mg uma vez ao dia com ou sem alimentos, a qualquer hora do dia.
Devido ao modo de ação específico da prucaloprida (estimulação da motilidade propulsiva), não se prevê que uma dose diária superior a 2 mg conduza a um aumento da eficácia.
Se a prucaloprida uma vez ao dia não for eficaz após 4 semanas de tratamento, o paciente precisará ser reexaminado e o benefício de continuar o tratamento deve ser avaliado.
A eficácia da prucaloprida foi demonstrada em estudos duplo-cegos controlados com placebo com duração até três meses.A eficácia para além de três meses não foi demonstrada em estudos controlados com placebo (ver secção 5.1). Em caso de tratamento prolongado, o benefício deve ser reavaliado em intervalos regulares.
Populações especiais
Idoso (> 65 anos): Comece com 1 mg uma vez ao dia (ver seção 5.2); se necessário, a dose pode ser aumentada até 2 mg uma vez por dia.
Pacientes com insuficiência renal: A dose para doentes com compromisso renal grave (TFG 2) é de 1 mg uma vez por dia (ver secções 4.3 e 5.2). Não é necessário ajuste de dose em pacientes com insuficiência renal leve a moderada.
Pacientes que sofrem de insuficiência hepática: Doentes com compromisso hepático grave (Child-Pugh classe C) começam com uma dose de 1 mg uma vez ao dia, que pode ser aumentada para 2 mg se necessário para melhorar a eficácia e se a dose de 1 mg for bem tolerada (ver secções 4.4 e 5.2 ) Não é necessário ajuste de dose em pacientes com insuficiência hepática leve a moderada.
População pediátrica: O Resolor não deve ser utilizado em crianças e adolescentes com menos de 18 anos (ver secção 5.1).
Método de administração
Uso oral.
04.3 Contra-indicações
• Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na secção 6.1.
• Insuficiência renal que requer diálise.
• Perfuração ou obstrução intestinal devido a distúrbios estruturais ou funcionais da parede intestinal, íleo obstrutivo, condições inflamatórias graves do trato intestinal, como doença de Crohn e colite ulcerativa, bem como megacólon / megareto tóxico.
04.4 Advertências especiais e precauções adequadas de uso
A principal via de eliminação da prucaloprida é a excreção renal (ver secção 5.2) .Recomenda-se uma dose de 1 mg em indivíduos com compromisso renal grave (ver secção 4.2).
O Resolor deve ser prescrito com precaução a doentes com compromisso hepático grave (Child-Pugh classe C), porque os dados sobre a utilização nestes doentes são limitados (ver secção 4.2).
A segurança e eficácia do Resolor para utilização em doentes com doença concomitante grave e clinicamente instável (por exemplo, doença cardiovascular ou pulmonar, doenças neurológicas ou psiquiátricas, cancro ou SIDA e outras doenças endócrinas) não foram estabelecidas em ensaios clínicos controlados. O Resolor deve ser prescrito com cautela a pacientes com essas condições, particularmente quando usado em pacientes com histórico de arritmias ou doença cardiovascular isquêmica.
Em caso de diarreia grave, a eficácia dos contraceptivos orais pode ser reduzida; portanto, o uso de um método contraceptivo adicional é recomendado para prevenir a possível ineficácia da contracepção oral (ver informações sobre a prescrição de contraceptivos orais).
Os comprimidos contêm lactose. Os doentes com problemas hereditários raros de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glucose-galactose não devem tomar este medicamento.
04.5 Interações com outros medicamentos e outras formas de interação
A prucaloprida tem um baixo potencial de interação farmacocinética. É amplamente excretado na forma inalterada na urina (aproximadamente 60% da dose) e no metabolismo em vitro é muito lento.
A prucaloprida não inibiu atividades específicas do CYP450 nos estudos em vitro em microssomas de fígado humano, em concentrações terapeuticamente relevantes.
Embora a prucaloprida possa ser um substrato fraco para a glicoproteína-P (gp-P), não é um inibidor da gp-P em concentrações clinicamente relevantes.
Efeitos da prucaloprida na farmacocinética de outros medicamentos
Houve um aumento de 30% nas concentrações plasmáticas de eritromicina durante o tratamento concomitante com prucaloprida. O mecanismo por trás dessa interação não é claro.
A prucaloprida não teve efeitos clinicamente relevantes na farmacocinética da varfarina, digoxina, álcool e paroxetina ou contraceptivos orais.
Efeitos de outros medicamentos na farmacocinética da prucaloprida
O cetoconazol (200 mg duas vezes ao dia), um inibidor potente do CYP3A4 e da gp-P, aumentou a exposição sistêmica à prucaloprida em aproximadamente 40%. Este efeito é muito pequeno para ser clinicamente relevante. Interações de significância semelhante. Podem ser esperadas com outro P potente inibidores -gp, tais como verapamil, ciclosporina A e quinidina.
Doses terapêuticas de probenecida, cimetidina, eritromicina e paroxetina não afetaram a farmacocinética da prucaloprida.
04.6 Gravidez e lactação
Mulheres com potencial para engravidar
Mulheres com potencial para engravidar devem usar métodos anticoncepcionais eficazes durante o tratamento com prucaloprida.
Gravidez
A experiência com a prucaloprida na gravidez é limitada. Foram observados casos de aborto espontâneo em estudos clínicos, embora na presença de outros fatores de risco, a relação com a prucaloprida seja desconhecida. Os estudos em animais não indicam efeitos diretos ou prejudiciais. Efeitos indiretos na gravidez, desenvolvimento embrionário / fetal, parto ou desenvolvimento pós-natal (ver secção 5.3). Resolor não é recomendado na gravidez.
Hora da alimentação
A prucaloprida é excretada no leite materno. No entanto, com doses terapêuticas de Resolor, não são esperados efeitos em recém-nascidos / lactentes amamentados. Na ausência de dados humanos, o uso de Resolor não é recomendado durante a lactação.
Fertilidade
Os estudos em animais indicam que não há efeito na fertilidade masculina ou feminina.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
O Resolor pode prejudicar ligeiramente a capacidade de conduzir e utilizar máquinas uma vez que foram observadas tonturas e fadiga em estudos clínicos, particularmente durante o primeiro dia de tratamento (ver secção 4.8).
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
Foi realizada uma análise integrada de 17 estudos duplo-cegos controlados por placebo nos quais o Resolor foi administrado por via oral a aproximadamente 3.300 pacientes com constipação crônica. Destes pacientes, mais de 1.500 tomaram Resolor na dose recomendada de 2 mg por dia, enquanto aproximadamente 1.360 foram tratados com 4 mg de prucaloprida por dia. As reações adversas mais frequentemente notificadas associadas ao tratamento com Resolor 2 mg são cefaleia (17,8%) e sintomas gastrointestinais (dor abdominal (13,7%), náuseas (13,7%) e diarreia (12,0%)). As reações adversas ocorrem principalmente no início da terapia e geralmente desaparecem alguns dias após a continuação do tratamento. Outras reações adversas foram ocasionalmente relatadas. A maioria dos eventos adversos foram de intensidade "leve a moderada".
Lista tabelada de reações adversas
As seguintes reações adversas foram notificadas em ensaios clínicos controlados na dose recomendada de 2 mg com frequências correspondentes a: muito frequentes (≥ 1/10), frequentes (≥ 1/100,
Descrição de algumas reações adversas
Após o primeiro dia de tratamento, as reações adversas mais comuns foram notificadas com frequência semelhante (diferença na incidência de não mais de 1% entre prucaloprida e placebo) durante a terapia com Resolor e durante o tratamento com placebo, com exceção de náuseas e diarreia que continuou a ocorrer com maior frequência durante a terapêutica com Resolor, embora menos pronunciada (diferenças na incidência entre Resolor e placebo de 1,3% (náuseas) e 3,4% (diarreia), respectivamente).
Palpitações foram relatadas em 0,7% dos pacientes tratados com placebo, 0,9% dos pacientes tratados com 1 mg de prucaloprida, 0,9% dos pacientes tratados com 2 mg de prucaloprida e 1,9% dos pacientes tratados com 4 mg de prucaloprida. A maioria dos pacientes continuou a tomar prucaloprida. Os pacientes devem discutir com o médico assistente, o novo aparecimento de palpitações, bem como o aparecimento de novos sintomas.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas que ocorram após a autorização do medicamento é importante, uma vez que permite a monitorização contínua da relação benefício / risco do medicamento.Os profissionais de saúde são convidados a notificar quaisquer suspeitas de reações adversas através da Agência Italiana de Medicamentos. . Site: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdose
Em um estudo em voluntários saudáveis, o tratamento com prucaloprida foi bem tolerado quando administrado em um regime crescente de até 20 mg uma vez ao dia (10 vezes a dose terapêutica recomendada). Uma sobredosagem pode resultar em sintomas que "amplificam os efeitos farmacodinâmicos conhecidos da prucaloprida e incluem cefaleias, náuseas e diarreia. Não existe tratamento específico para a sobredosagem com Resolor. Caso ocorra uma sobredosagem, o doente deve ser tratado sintomaticamente e deve ser tratado". devem ser tomadas medidas conforme necessário. A perda excessiva de fluidos causada por diarreia ou vômito pode exigir a correção dos desequilíbrios eletrolíticos.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: outros laxantes, código ATC: A06AX05.
Mecanismo de ação
A prucaloprida é uma di-hidrobenzofuranocarboxamida com atividade procinética gastrointestinal.A prucaloprida é um agonista seletivo do receptor da serotonina (5-HT4) de alta afinidade, o que provavelmente explica seus efeitos procinéticos. Em vitro uma "afinidade para outros receptores foi encontrada apenas em concentrações que excedem sua afinidade para o receptor 5-HT4 em pelo menos 150 vezes. Em ratos, prucaloprida na Vivo, em doses acima de 5 mg / kg (30-70 vezes a exposição clínica e além), induz hiperprolactinemia causada por uma ação antagonista contra o receptor D2.
Em cães, a prucaloprida altera os padrões de motilidade do cólon por meio da estimulação do receptor 5-HT4 da serotonina: estimula a motilidade do cólon proximal, melhora a motilidade gastroduodenal e acelera o esvaziamento gástrico retardado. A prucaloprida também induz contrações migratórias gigantes. Estes são equivalentes aos movimentos de massa do cólon em humanos e fornecem a principal força motriz para a defecação. Em cães, os efeitos observados no trato gastrointestinal são sensíveis ao bloqueio com antagonistas seletivos do receptor 5-HT4, demonstrando assim que os efeitos observados são exercida pela ação seletiva nos receptores 5-HT4.
Esses efeitos farmacodinâmicos da prucaloprida foram confirmados em humanos por manometria usada em indivíduos com constipação crônica em um estudo randomizado, aberto e cruzado com avaliação cega dos efeitos da prucaloprida 2 mg e um laxante osmótico na motilidade do cólon com base no número de contrações do cólon que se propagam em grande amplitude (HAPC, contrações de propagação de alta amplitude, também conhecidas como contrações migrantes gigantes). Em comparação com os tratamentos para constipação osmótica, a estimulação procinética produzida pela prucaloprida aumentou a motilidade do cólon na extensão expressa pelo número de HAPCs nas 12 horas após a administração do produto experimental. O benefício ou relevância clínica deste mecanismo de ação em relação a outros laxantes não foi estudado.
Eficácia clínica e segurança
População adulta
A eficácia do Resolor foi demonstrada em três estudos multicêntricos, randomizados, duplo-cegos, de 12 semanas, controlados por placebo em indivíduos com constipação crônica (n = 1.279 tratados com Resolor, 1.124 mulheres, 155 homens). As doses de Resolor avaliadas em cada um desses três estudos foi de 2 mg e 4 mg uma vez ao dia. O objetivo primário de eficácia foi a porcentagem (%) de indivíduos que alcançaram a normalização dos movimentos intestinais, definida como uma média de três ou mais movimentos intestinais espontâneos e completos (Evacuações intestinais completas espontâneas, SCBM) por semana durante o período de tratamento de 12 semanas.
A porcentagem de pacientes do sexo feminino para as quais os laxantes não proporcionaram alívio adequado, tratadas com a dose recomendada de 2 mg de Resolor (n = 458), que alcançaram uma média de ≥ 3 SCBMs por semana foi de 31,0% (semana 4) e 24,7 % (semana 12), versus 8,6% (semana 4) e 9,2% (semana 12) no grupo de placebo. Uma melhora clinicamente significativa em ≥1 SCBM por semana, o endpoint secundário de eficácia mais importante, foi alcançada em 51,0% (semana 4) e 44,2% (semana 12) dos indivíduos tratados com 2 mg de Resolor, versus 21,7% (semana 4) e 22,6% (semana 12) de placebo.
O efeito do Resolor nas evacuações espontâneas (Movimentos intestinais espontâneos, SBM) também foi estatisticamente superior ao placebo na proporção de pacientes que tiveram um aumento ≥1 SBM / semana durante o período de tratamento de 12 semanas. Na semana 12, 68,3% dos pacientes tratados com Resolor 2 mg tiveram um aumento médio de ≥1 SBM / semana, em comparação com 37,0% dos pacientes tratados com placebo (p
Em todos os três estudos, o tratamento com Resolor também resultou em melhorias significativas nas avaliações de uma gama de sintomas patológicos específicos e validados (PAC-SYM, avaliação dos sintomas de constipação do paciente), que incluem sintomas abdominais (inchaço, desconforto, dor e cólicas), defecadores (evacuações incompletas, alarmes falsos, esforço, dureza excessiva das fezes, volume insuficiente de fezes) e retal (evacuações dolorosas, queimação, sangramento / lacrimejamento), avaliados na semana 4 e na semana 12. Na semana 4, a porcentagem de pacientes com ≥ 1 melhora da linha de base nas subescalas do PAC-SYM de sintomas abdominais, defecatórios e retais foi de 41,3%, 41,6% e 31,3%, respectivamente, em pacientes tratados com Resolor 2 mg, em comparação com 26,9%, 24,4% e 22,9% em placebo- pacientes tratados. Resultados semelhantes foram observados na Semana 12: 43,4%, 42,9% e 31,7%, respectivamente, em pacientes tratados com Resolor 2 mg, em comparação com 26,9%, 27,2% e 23,4% em pacientes tratados com placebo (p
Em ambas as avaliações, na semana 4 e na semana 12, também foi observado benefício significativo em relação a uma série de parâmetros relativos à qualidade de vida, como nível de satisfação com o tratamento, hábitos e preocupações intestinais, desconforto e aspectos físicos e psicossociais. desconforto. Na semana 4, a porcentagem de pacientes com melhora ≥1 da linha de base na subescala Avaliação da Constipação Subjetiva - Qualidade de Vida (PAC-QOL) foi de 47,7% em pacientes tratados com Resolor 2 mg, em comparação com 20,2% em pacientes tratados com placebo. Resultados semelhantes foram observados na Semana 12: 46,9% em pacientes tratados com Resolor 2 mg, respectivamente, em comparação com 19,0% em pacientes tratados com placebo (p
Além disso, a eficácia, segurança e tolerabilidade do Resolor em pacientes do sexo masculino com constipação crônica foram avaliadas em um estudo multicêntrico, randomizado, duplo-cego e controlado por placebo de 12 semanas (N = 370). O desfecho primário do estudo foi atendido: uma proporção maior estatisticamente significativa de indivíduos no grupo Resolor (37,9%) tinha um SCMB médio semanal ≥ 3, em comparação com os indivíduos no grupo placebo (17,7%) (p
Estudo de longo prazo
A eficácia e a segurança do Resolor em pacientes (idade ≥18 anos) com constipação crônica foram avaliadas em um estudo multicêntrico, randomizado, duplo-cego, controlado por placebo de 24 semanas (N = 361). De pacientes com uma frequência semanal média de evacuações espontâneas e completas (SCBM) ≥3 (respondedores) durante a fase de tratamento duplo-cego de 24 semanas não foi estatisticamente diferente (p = 0,367) entre os grupos de tratamento com Resolor (25,1%) e placebo (20,7%). A diferença entre os grupos de tratamento da frequência média semanal de SCBM ≥3 não foi estatisticamente significativa para as Semanas 1 a 12, um resultado em contraste com os outros 5 estudos multicêntricos, randomizados, duplo-cegos e controlados por placebo. 12 semanas de duração que demonstraram a eficácia de prucaloprida em pacientes adultos no mesmo período de avaliação, portanto, o estudo é considerado inconclusivo no que diz respeito à eficácia. No entanto, a totalidade dos dados, incluindo os outros estudos duplo-cegos, controlados por placebo, de 12 semanas apoiam a eficácia do Resolor. O perfil de segurança do Resolor observado neste estudo de 24 semanas está em linha com aquele. Estudos de 12 semanas.
Resolor não mostrou nenhum fenômeno de rebote e não era viciante.
Estudo aprofundado sobre QT
Um estudo QT completo foi realizado para avaliar os efeitos do Resolor no intervalo QT em doses terapêuticas (2 mg) e supraterapêuticas (10 mg) e os resultados foram comparados com os efeitos do placebo e de um controle positivo. demonstraram diferenças significativas entre o Resolor usado em ambas as doses e o placebo com base nas medições médias do QT e em uma "análise de valor anormal". Isso confirmou os resultados de dois estudos QT controlados com placebo. Em estudos clínicos duplo-cegos, a incidência de eventos adversos relacionados ao QT e arritmias ventriculares foi baixa e comparável à do grupo placebo.
População pediátrica
A eficácia e segurança do Resolor em pacientes pediátricos (idades de 6 meses - 18 anos) com constipação funcional foram avaliadas em um estudo duplo-cego controlado por placebo de 8 semanas (N = 213), seguido por um comparador aberto estudo controlado (polietilenoglicol 4000) de 16 semanas com duração de até 24 semanas (N = 197). Crianças com peso ≤50 kg receberam uma dose inicial de 0,04 mg / kg / dia aumentada gradualmente entre 0,02 e 0,06 mg / kg / dia ( para um máximo de 2 mg / dia) de Resolor solução oral ou placebo correspondente.Crianças com peso> 50 kg receberam 2 mg / dia de comprimidos de Resolor ou placebo correspondente.
A resposta ao tratamento foi definida como uma média de ≥3 evacuações espontâneas (SBM) por semana e um número médio de episódios de incontinência fecal ≤1 a cada 2 semanas. Os resultados do estudo não mostraram diferenças. Em termos de eficácia entre Resolor e placebo: taxas de resposta foram 17% e 17,8%, respectivamente (P = 0,9002). Resolor foi geralmente bem tolerado. A incidência de indivíduos com pelo menos 1 início de evento adverso durante o tratamento (TEAE) foi semelhante entre o grupo Resolor (69,8%) e o grupo placebo (60,7%). Globalmente, o perfil de segurança do Resolor nas crianças foi igual ao dos adultos.
05.2 "Propriedades farmacocinéticas
Absorção
A prucaloprida é rapidamente absorvida; após uma dose oral única de 2 mg, a Cmax foi atingida em 2-3 horas. A biodisponibilidade oral absoluta é> 90%. A ingestão concomitante de alimentos não afeta a biodisponibilidade oral da prucaloprida.
Distribuição
A prucaloprida é amplamente distribuída e tem um volume de distribuição em estado estacionário (Vdss) de 567 litros. A ligação da prucaloprida às proteínas é de cerca de 30%.
Biotransformação
O metabolismo não é a principal via de eliminação da prucaloprida. Em vitro, o metabolismo do fígado humano é muito lento e apenas uma quantidade mínima de metabólitos é encontrada. Num estudo humano de dose oral com prucaloprida marcada radioactivamente, foram encontradas pequenas quantidades de sete metabolitos na urina e nas fezes. O metabolito quantitativamente mais representado na excreção, R107504, foi responsável por 3,2% e 3,1%, respectivamente. Da dose na urina e nas fezes. Outros metabólitos identificados e quantificados na urina e nas fezes foram R084536 (formado por N-desalquilação), correspondendo a 3% da dose e os produtos de hidroxilação (3% da dose) e N-oxidação (2% da dose). A substância ativa inalterada constituiu aproximadamente 92-94% da radioatividade total no plasma. R107504, R084536 e R104065 (formado por O-desmetilação) foram identificados como metabolitos plasmáticos menores.
Eliminação
Uma grande fração da substância ativa é eliminada inalterada (60-65% da dose administrada na urina e cerca de 5% nas fezes). A excreção renal da prucaloprida inalterada envolve filtração passiva e secreção ativa. A depuração plasmática da prucaloprida é em média 317 ml / minuto. Sua meia-vida terminal é de aproximadamente um dia. O estado de equilíbrio é atingido em três a quatro dias. Com tratamento uma vez ao dia com 2 mg prucaloprida, as concentrações plasmáticas em estado estacionário flutuam entre os valores mínimo e máximo de 2,5 e 7 ng / ml, respectivamente. A razão de acumulação após uma única dose diária variou de 1, 9 e 2,3. A farmacocinética da prucaloprida é proporcional à dose tanto dentro faixa terapêutica e além (testado até 20 mg) .Prucaloprida administrada uma vez ao dia exibe cinética independente do tempo durante o tratamento prolongado.
Populações especiais
Farmacocinética populacional
Uma análise farmacocinética populacional mostrou que a depuração total aparente da prucaloprida estava correlacionada com a depuração da creatinina e que a idade, peso corporal, sexo ou raça não tiveram influência.
Cidadãos idosos
Após uma dose única diária de 1 mg, as concentrações plasmáticas máximas e a AUC da prucaloprida em idosos foram 26-28% mais elevadas do que em adultos jovens.Este efeito pode ser atribuído a uma diminuição da função renal em idosos.
Falência renal
Em comparação com indivíduos com função renal normal, as concentrações plasmáticas de prucaloprida após uma dose única de 2 mg foram em média 25% e 51% maiores em indivíduos com insuficiência renal leve, respectivamente (ClCR 50-79 ml / minutos). E moderada (ClCR25 -49 ml / minutos). Em indivíduos com compromisso renal grave (ClCR ≤ 24 ml / minutos), as concentrações plasmáticas foram 2,3 vezes os níveis encontrados em indivíduos saudáveis (ver secções 4.2 e 4.4).
Insuficiência Hepática
A eliminação não renal contribui com aproximadamente 35% para a eliminação total. Num pequeno estudo farmacocinético, a Cmax e a AUC da prucaloprida foram em média 10-20% mais elevadas em doentes com compromisso hepático moderado a grave do que em indivíduos saudáveis (ver secções 4.2 e 4.4).
05.3 Dados de segurança pré-clínica
Os dados não clínicos não revelam riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico, toxicidade reprodutiva e de desenvolvimento. Um grande número de estudos de segurança de medicamentos, realizados com particular atenção aos parâmetros cardiovasculares, não mostraram alterações relevantes nos parâmetros hemodinâmicos e derivados do ECG (QTc), exceto por um aumento modesto na frequência cardíaca e pressão arterial observada em porcos anestesiados após administração intravenosa administração, e um aumento da pressão arterial em cães conscientes após administração intravenosa em bólus, o que, no entanto, não foi observado em cães anestesiados ou após administração oral em cães nos quais níveis plasmáticos semelhantes são alcançados. Um estudo de toxicidade subcutânea neonatal / juvenil conduzido em ratos de 7 a 55 dias de idade mostrou um NOAEL de 100 mg / kg / dia. As taxas de exposição determinadas com base na AUC0-24h no NOAEL em comparação com aquelas detectadas em pacientes pediátricos (tratados com aproximadamente 0,04 mg / kg / dia) foram incluídas na faixa de 21 a 71, garantindo assim margens de segurança adequadas para o ambiente clínico dose.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Miolo de cornpress
Lactose monohidratada
Celulose microcristalina
Dióxido de sílica coloidal
Estearato de magnesio
Revestimento de comprimido
Hipromelose
Lactose monohidratada
Triacetina
Dióxido de titânio (E171)
Macrogol
06.2 Incompatibilidade
Não é relevante.
06.3 Período de validade
4 anos.
06.4 Precauções especiais de armazenamento
Conservar no blister original para proteger da humidade.
06.5 Natureza da embalagem primária e conteúdo da embalagem
Blisters de dose unitária de alumínio / alumínio perfurados (calendário) contendo 7 comprimidos. Cada embalagem contém 7 x 1, 14 x 1, 28 x 1 ou 84 x 1 comprimidos revestidos por película.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio
Sem instruções especiais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Shire Pharmaceuticals Ireland Limited
5 Riverwalk
Citywest Business Campus
Dublin 24
Irlanda
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU / 1/09/581/001 (28 comprimidos)
EU / 1/09/581/003 (7 comprimidos)
EU / 1/09/581/005 (14 comprimidos)
EU / 1/09/581/007 (84 comprimidos)
041016015
041016027
041016041
041016066
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 15 de outubro de 2009
Data da renovação mais recente: 06 de junho de 2014
10.0 DATA DE REVISÃO DO TEXTO
05/2015