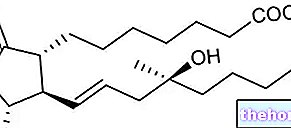
Ingredientes ativos: infliximabe
Remicade 100 mg pó para concentrado para solução para perfusão
Por que o Remicade é usado? Para que serve?
Remicade contém uma substância ativa chamada infliximab. O infliximabe é uma proteína de origem humana e animal (de camundongo).
Remicade pertence a um grupo de medicamentos denominados “bloqueadores do TNF”. É usado em adultos para o tratamento das seguintes doenças inflamatórias:
- Artrite reumatóide
- Artrite psoriática
- Espondilite anquilosante (doença de Bechterew)
- Psoríase.
Remicade também é usado em adultos e crianças de 6 anos de idade ou mais para:
- Doença de crohn
- Colite ulcerativa.
Remicade atua bloqueando a ação de uma proteína denominada “fator de necrose tumoral alfa” (TNFα) .Esta proteína está envolvida nos processos inflamatórios do organismo e, ao bloqueá-la, é possível reduzir a inflamação no organismo.
Artrite reumatóide
A artrite reumatóide é uma doença inflamatória das articulações. Se tiver artrite reumatóide, será inicialmente tratado com outros medicamentos. Se não responder adequadamente a estes medicamentos, será tratado com Remicade em combinação com outro medicamento denominado metotrexato para:
- Reduza os sinais e sintomas da doença,
- Retardar a progressão dos danos às articulações,
- Melhore a função física.
Artrite psoriática
A artrite psoriática é uma doença inflamatória das articulações, geralmente acompanhada de psoríase. Se tiver artrite psoriática, será tratado primeiro com outros medicamentos. Se não responder de forma adequada a estes medicamentos, será tratado com Remicade para:
- Reduza os sinais e sintomas da doença,
- Retardar a progressão dos danos às articulações,
- Melhore a função física.
Espondilite anquilosante (doença de Bechterew)
A espondilite anquilosante é uma doença inflamatória da coluna vertebral. Se tiver espondilite anquilosante, primeiro será tratado com outros medicamentos. Se você não responder adequadamente a esses medicamentos, você será tratado com Remicade para:
- Reduza os sinais e sintomas da doença,
- Melhore a função física.
Psoríase
A psoríase é uma doença inflamatória da pele. Se tem psoríase em placas moderada a grave, será tratado primeiro com outros medicamentos ou outros tratamentos, como fototerapia. Se não responder adequadamente a esses medicamentos ou tratamentos, você será tratado com Remicade para reduzir os sinais e sintomas da sua doença.
Colite ulcerativa
A colite ulcerosa é uma doença inflamatória do intestino. Se tiver colite ulcerosa, será tratado primeiro com outros medicamentos. Se não responder de forma adequada a estes medicamentos, ser-lhe-á administrado Remicade para tratar a doença.
Doença de crohn
A doença de Crohn é uma doença inflamatória intestinal. Se tiver doença de Crohn, será tratado com outros medicamentos primeiro. Se não responder adequadamente a estes medicamentos, será tratado com Remicade para: • Tratar a doença de Crohn ativa • Reduzir o número de aberturas anormais (fístulas) entre o intestino e a pele, para as quais outros medicamentos ou cirurgias se mostraram inadequados.
Contra-indicações Quando Remicade não deve ser usado
Você não deve receber Remicade se:
- tem alergia ao infliximab (substância ativa do Remicade) ou a qualquer outro componente deste medicamento (listados na seção 6)
- você é alérgico (hipersensível) a proteínas de camundongo
- tem tuberculose (TB) ou uma "outra infecção grave, como pneumonia ou sepse
- tem "insuficiência cardíaca moderada ou grave".
Não tome Remicade se alguma das condições acima se aplicar a si. Se você não tem certeza, converse com seu médico antes de receber Remicade
Precauções de uso O que você precisa saber antes de tomar Remicade
Fale com o seu médico antes de receber Remicade se tiver:
Remicade recebido anteriormente
- Informe o seu médico se já fez tratamento com Remicade no passado e se está a reiniciar o tratamento com Remicade.
Se parou de tomar Remicade por mais de 16 semanas, existe um risco aumentado de reações alérgicas quando reiniciar Remicade.
Infecções
Informe o seu médico se tiver uma "infecção, mesmo que seja ligeira, antes de lhe ser administrado Remicade
- Informe o seu médico antes de receber Remicade se você morou ou viajou para uma "área onde infecções chamadas histoplasmose, coccidioidomicose ou blastomicose são comuns. Essas infecções são causadas por tipos específicos de fungos que podem afetar os pulmões ou outras partes do corpo corpo
- Pode ficar mais sujeito a infecções quando tratado com Remicade. Se você tem 65 anos ou mais, você tem um risco maior
- Estas infecções podem ser graves e incluir tuberculose, infecções causadas por vírus, fungos ou bactérias ou outras infecções oportunistas e sépsis que podem, em casos raros, ser fatais.
Informe o seu médico imediatamente se tiver quaisquer sintomas de infecção durante o tratamento com Remicade. Os sintomas incluem febre, tosse, sintomas semelhantes aos da gripe, mal-estar, pele vermelha ou muito quente, feridas ou problemas dentários. O seu médico pode recomendar uma interrupção temporária do Remicade.
Tuberculose (TB)
- É muito importante que informe o seu médico se alguma vez teve tuberculose ou se esteve em contacto próximo com pessoas que já tiveram ou têm tuberculose
- Seu médico fará testes para ver se você tem tuberculose. Foram notificados alguns casos de tuberculose em doentes tratados com Remicade, em raras ocasiões, mesmo em doentes que tinham sido tratados com medicamentos para a tuberculose. O médico registrará esses testes no Cartão de Alerta do Paciente
- Se o seu médico achar que você está em risco de contrair tuberculose, você pode ser tratado com medicamentos para a tuberculose antes de receber Remicade.
Informe imediatamente o seu médico se notar quaisquer sinais de tuberculose enquanto estiver a tomar Remicade. Os sinais incluem tosse persistente, perda de peso, sensação de cansaço, febre, suores noturnos.
Vírus da hepatite B (HBV)
- Informe o seu médico se você é portador ou tem ou já teve hepatite B antes de receber Remicade
- Informe o seu médico se você acha que pode estar em risco de contrair hepatite B
- O médico deve avaliar se você tem hepatite B? O tratamento com bloqueadores do TNF, como o Remicade, pode causar a reativação do vírus da hepatite B em pacientes com esse vírus, o que em alguns casos pode causar a morte.
Problemas cardíacos
- Informe o seu médico se você tiver algum problema de coração, como insuficiência cardíaca leve
- O seu médico monitorará de perto a sua função cardíaca.
Informe imediatamente o seu médico se notar sinais novos ou agravamento de insuficiência cardíaca durante o tratamento com Remicade. Os sinais incluem falta de ar ou inchaço dos pés.
Câncer e linfoma
- Informe o seu médico se você tem ou já teve linfoma (um tipo de câncer no sangue) ou outros tipos de câncer antes de receber Remicade
- Pacientes com artrite reumatoide grave que sofrem dessa doença há muito tempo podem ter um risco maior do que a média de desenvolver linfoma.
- Crianças e adultos que tomam Remicade podem ter um risco aumentado de desenvolver linfoma ou outro tipo de câncer.
- Alguns pacientes que foram tratados com bloqueadores de TNF, incluindo Remicade, desenvolveram um tipo raro de câncer chamado Linfoma Hepatosplênico de Células T. A maioria desses pacientes eram adolescentes ou adultos jovens do sexo masculino e a maioria tinha Crohn ou colite ulcerosa. Esse tipo de câncer geralmente é letal. Quase todos os pacientes também foram tratados com medicamentos chamados azatioprina ou 6-mercaptopurina, além de bloqueadores de TNF.
- Alguns pacientes tratados com infliximabe desenvolveram certos tipos de câncer de pele. Se sentir qualquer tipo de alteração na aparência da pele ou crescimentos na pele durante ou após a terapia, informe o seu médico.
Doença pulmonar ou tabagismo pesado
- Informe o seu médico se você tem uma doença pulmonar chamada doença pulmonar obstrutiva crônica (DPOC) ou se você fuma muito antes de tomar Remicade
- Pacientes com DPOC e fumantes inveterados podem apresentar risco aumentado de câncer quando tratados com Remicade.
Doença do sistema nervoso
- Informe o seu médico se você tem ou já teve um problema de sistema nervoso antes de receber Remicade. Isso inclui esclerose múltipla, síndrome de Guillain-Barré, ataques ou um diagnóstico de "neurite óptica".
Informe o seu médico imediatamente se notar sintomas de uma doença dos nervos enquanto estiver a tomar Remicade. Os sinais incluem alterações na visão, fraqueza nos braços e pernas, dormência ou formigamento em qualquer parte do corpo.
Aberturas anormais da pele
- Informe o seu médico se tiver quaisquer aberturas anormais na pele (fístulas) antes de lhe ser administrado Remicade.
Vacinas
- Informe o seu médico se você foi vacinado recentemente ou planeja ser vacinado
- Você não deve receber vacinas durante o tratamento com Remicade
- Algumas vacinas podem causar infecções. Se você recebeu Remicade durante a gravidez, seu bebê pode ter um risco aumentado de contrair esta infecção por aproximadamente seis meses após a última dose recebida durante a gravidez. É importante informar o seu pediatra e outros profissionais de saúde sobre o uso de Remicade para que possam decidir quando seu filho deve receber qualquer vacina.
Agentes terapêuticos infecciosos
- Fale com o seu médico se você tomou recentemente ou está planejando fazer o tratamento com um agente terapêutico infeccioso (como a instilação de BCG usada para tratar o câncer).
Operações ou procedimentos odontológicos
- Informe o seu médico se você vai fazer algum procedimento ou tratamento dentário
- Informe o cirurgião ou dentista que realiza o procedimento que você está sendo tratado com Remicade, mostrando o cartão de alerta do paciente.
Crianças e adolescentes
As informações acima também se aplicam a crianças e adolescentes. Além disso:
- alguns doentes crianças e adolescentes que tomaram medicamentos bloqueadores do TNF, como o Remicade, desenvolveram cancros, incluindo tipos invulgares, que por vezes podem ser fatais.
- Em comparação com os adultos, mais crianças que tomaram Remicade desenvolveram infecções
- As crianças devem receber as vacinas recomendadas antes de iniciar o tratamento com Remicade.
Se não tem a certeza se alguma das condições anteriores se aplica a si, contacte o seu médico antes de lhe ser administrado Remicade.
Interações Quais medicamentos ou alimentos podem alterar o efeito do Remicade
Pacientes com doenças inflamatórias já tomam remédios para tratar a doença. Esses medicamentos podem causar efeitos colaterais. O seu médico irá aconselhá-lo sobre os outros medicamentos que deve continuar a tomar enquanto estiver a ser tratado com Remicade.
Informe o seu médico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo quaisquer outros medicamentos para tratar a doença de Crohn, colite ulcerosa, artrite reumatóide, espondilite anquilosante, artrite psoriática ou psoríase ou medicamentos que obtenha sem receita, como vitaminas e ervas remédios.
Em particular, informe o seu médico se você estiver usando algum destes medicamentos:
- Medicamentos que afetam o sistema imunológico
- Kineret (anakinra). Remicade e Kineret não devem ser administrados juntos
- Orencia (abatacept). Remicade e Orencia não devem ser administrados juntos.
Se não tem a certeza se alguma das condições anteriores se aplica a si, contacte o seu médico antes de lhe ser administrado Remicade.
Avisos É importante saber que:
Gravidez, amamentação e fertilidade
- Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico antes de tomar este medicamento. Remicade não é recomendado durante a gravidez
- Deve evitar engravidar durante o tratamento com Remicade e durante pelo menos 6 meses após interromper o tratamento.Certifique-se de que está a utilizar métodos contracetivos adequados durante este período.
- Não amamente enquanto estiver sendo tratado com Remicade ou por 6 meses após o último tratamento com Remicade
- Se você recebeu Remicade durante a gravidez, seu bebê pode ter um risco aumentado de contrair uma infecção. É importante informar o seu pediatra e outros profissionais de saúde sobre o uso de Remicade antes de seu bebê receber qualquer vacina (para mais informações, consulte a seção sobre vacinações )
Condução e utilização de máquinas
É improvável que Remicade afete a sua capacidade de conduzir ou utilizar máquinas. Se se sentir cansado ou indisposto após o tratamento com Remicade, não deve conduzir ou utilizar quaisquer ferramentas ou máquinas.
Dose, Método e Tempo de Administração Como usar Remicade: Posologia
Como Remicade é administrado
- Remicade ser-lhe-á administrado pelo seu médico ou enfermeiro
- O seu médico ou enfermeiro irá preparar a solução injetável de Remicade
- A solução de Remicade será injetada lentamente (ao longo de um período de 2 horas) em uma veia, geralmente no braço. Este procedimento é denominado "infusão intravenosa" ou gotejamento. Após o terceiro tratamento, o seu médico pode decidir dar-lhe Remicade durante um período de 1 hora
- Você será monitorado durante a administração de Remicade e por 1-2 horas depois.
Quanto Remicade é dado
- O seu médico irá determinar a dose (em mg) e o intervalo entre as doses de Remicade, que dependerá da sua doença, peso e resposta ao tratamento.
- A tabela abaixo mostra a frequência de administração deste medicamento.
Artrite reumatóide
A dose usual é de 3 mg para cada kg de peso corporal
Artrite psoriática, espondilite anquilosante (doença de Bechterew), psoríase, colite ulcerosa e doença de Crohn
A dose usual é de 5 mg por cada kg de peso corporal.
Uso em crianças e adolescentes
Remicade só deve ser utilizado em crianças com doença de Crohn ou colite ulcerosa. Essas crianças devem ter 6 anos de idade ou mais.
Overdose O que fazer se você tiver tomado muito Remicade
Se você receber mais Remicade do que precisa
Como este medicamento é administrado pelo seu médico ou enfermeiro, é improvável que receba uma quantidade excessiva. Não há efeitos colaterais conhecidos da sobredosagem de Remicade.
Se você esquecer ou perder uma infusão de "Remicade"
Se você esquecer ou faltar a uma consulta para administrar Remicade, marque outra consulta o mais rápido possível.
Caso ainda tenha dúvidas sobre o uso deste medicamento, pergunte ao seu médico
Efeitos colaterais Quais são os efeitos colaterais do Remicade
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham. A maioria desses efeitos são leves a moderados. No entanto, alguns pacientes podem apresentar efeitos colaterais graves e exigir tratamento médico. Os efeitos colaterais também podem ocorrer após o término do tratamento com Remicade.
Informe o seu médico imediatamente se notar algum dos seguintes efeitos colaterais:
- Sinais de uma reação alérgica, como inchaço da face, lábios, boca ou garganta que pode causar dificuldade em engolir ou respirar, erupção na pele, urticária, inchaço das mãos, pés ou tornozelos. A reação alérgica pode ocorrer 2 horas após a injeção ou mais tarde. Outros sinais de uma reação alérgica que podem ocorrer até 12 dias após a injeção incluem dores musculares, febre, dores nas articulações ou na mandíbula, garganta ou garganta inflamadas.
- Sinais de um problema cardíaco, como falta de ar, inchaço dos pés ou alterações no batimento cardíaco
- Sinais de uma infecção (incluindo tuberculose), como febre, sensação de cansaço, tosse (persistente), falta de ar, sintomas semelhantes aos da gripe, perda de peso, suores noturnos, diarreia, feridas, problemas dentários ou queimação ao urinar
- Sinais de um problema pulmonar, como tosse, dificuldade em respirar ou aperto no peito
- Sinais de problemas neurológicos (incluindo problemas oculares), como convulsões, formigamento ou dormência em qualquer parte do corpo, fraqueza nos braços ou pernas, alterações na visão, como visão dupla ou outros problemas oculares
- Sinais de um problema de fígado, como pele ou olhos amarelados, urina castanho-escura ou dor na parte superior direita do estômago, febre
- Sinais de um distúrbio do sistema imunológico, chamado lúpus, como dores nas articulações ou erupções nas bochechas ou braços, áreas sensíveis ao sol
- Sinais de diminuição do número de células sanguíneas, como febre persistente, hemorragia ou nódoas negras com maior frequência ou aparência pálida.
Se você notar algum dos sintomas descritos acima, informe o seu médico imediatamente.
Efeitos colaterais muito comuns (afetam mais de 1 em 10 pacientes)
- Dor de estômago, mal-estar
- Infecções virais como herpes ou gripe
- Infecções do trato respiratório superior, como sinusite
- Dor de cabeça
- Efeito indesejável devido à infusão
- Dor.
Efeitos colaterais comuns (afetam 1 a 10 usuários em 100)
- Alterações na função hepática, aumento das enzimas hepáticas (observadas em exames de sangue)
- Infecções dos pulmões ou do tórax, como bronquite ou pneumonia
- Dificuldades respiratórias ou dor ao respirar, dor no peito
- Sangramento no estômago ou intestinos, diarreia, indigestão, azia, prisão de ventre
- Erupção cutânea semelhante à urticária, erupção cutânea com comichão ou pele seca
- Problemas de equilíbrio ou tontura
- Febre, aumento da sudorese
- Problemas de circulação, como pressão alta ou baixa
- Hematomas, rubor ou sangramento nasal, pele quente e vermelha (vermelhidão)
- Sensação de cansaço ou fraqueza
- Infecções bacterianas, como infecção generalizada, abcesso ou infecção das camadas profundas da pele (celulite)
- Problemas de sangue, como anemia ou contagem baixa de glóbulos brancos
- Linfonodos aumentados
- Depressão, distúrbios do sono
- Problemas oculares, incluindo olhos vermelhos e infecções
- Batimento cardíaco rápido (taquicardia) ou palpitações
- Dor nas articulações, músculos ou costas
- Infecção do trato urinário
- Psoríase, problemas de pele como eczema e queda de cabelo
- Reações no local da injeção, como dor, inchaço, vermelhidão ou coceira
- Calafrios, acúmulo de fluido sob a pele causando inchaço
- Sensação de dormência ou formigamento.
Efeitos colaterais incomuns (afetam 1 a 10 usuários em 1.000)
- Suprimento insuficiente de sangue, inchaço de uma veia
- Problemas de pele como bolhas, verrugas, descoloração ou pigmentação anormal da pele ou lábios inchados
- Reações alérgicas graves (por exemplo, anafilaxia), distúrbio do sistema imunológico chamado lúpus, reações alérgicas a proteínas estranhas
- Feridas que demoram a cicatrizar
- Inchaço do fígado (hepatite) ou vesícula biliar (vesícula biliar), danos ao fígado
- Distração, irritabilidade, confusão, nervosismo
- Problemas oculares, incluindo visão turva ou reduzida, olhos inchados ou chiqueiros
- Insuficiência cardíaca nova ou agravada, frequência cardíaca lenta
- Desmaio
- Convulsões, distúrbios nervosos
- Perfuração intestinal ou bloqueio intestinal, dor de estômago ou cólicas
- Inchaço do pâncreas (pancreatite)
- Infecções fúngicas, como infecção por fungos
- Problemas pulmonares (como edema)
- Excesso de líquido ao redor dos pulmões (derrame pleural)
- Infecções renais
- Baixa contagem de plaquetas, número excessivo de leucócitos
- Infecções na vagina.
Efeitos colaterais raros (afetam 1 a 10 usuários em 10.000)
- Um tipo de câncer no sangue (linfoma)
- Fornecimento insuficiente de oxigênio aos órgãos através do sangue, problemas de circulação, como estreitamento de um vaso sanguíneo
- Inflamação da membrana que reveste o cérebro (meningite)
- Infecções devido a um sistema imunológico enfraquecido
- Infecção por hepatite B, se você já teve hepatite B no passado? Inchaço ou crescimento de tecidos anormais
- Inchaço dos pequenos vasos sanguíneos (vasculite)? Distúrbios imunológicos que podem afetar os pulmões, a pele e os gânglios linfáticos (como sarcoidose)
- Falta de interesse ou emoção
- Problemas graves de pele, como necrólise epidérmica tóxica, síndrome de Steven-Johnson ou eritema multiforme, problemas de pele como furúnculos
- Doenças graves do sistema nervoso, como mielite transversa, doença semelhante à esclerose múltipla, neurite óptica e síndrome de Guillain-Barré
- Fluido na membrana que reveste o coração (derrame pericárdico)
- Problemas pulmonares graves (como pneumonia intersticial)
- Melanoma (um tipo de câncer de pele).
Outros efeitos colaterais (a frequência é desconhecida)
- Câncer em crianças e adultos
- Um câncer de sangue raro que afeta principalmente pessoas jovens (linfoma hepatoesplênico de células T)
- Insuficiência Hepática
- Carcinoma de células de Merkel (um tipo de câncer de pele)
- Piora de uma condição chamada dermatomiosite (parece uma "erupção que acompanha a fraqueza muscular).
Efeitos colaterais adicionais em crianças e adolescentes
As crianças que tomaram Remicade para a doença de Crohn mostraram algumas diferenças nos efeitos colaterais em comparação com os adultos que tomaram Remicade para a doença de Crohn.
Os efeitos colaterais mais comuns em crianças foram: contagem baixa de glóbulos vermelhos (anemia), sangue nas fezes, contagem baixa de glóbulos brancos (leucopenia), rubor ou vermelhidão (ondas de calor), infecções virais, baixo número de neutrófilos (neutropenia) que são glóbulos brancos que combatem infecções, fraturas ósseas, infecções bacterianas e reações alérgicas do trato respiratório.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico, farmacêutico ou enfermeiro. Isto inclui quaisquer efeitos secundários possíveis não listados neste folheto. Também pode comunicar os efeitos secundários diretamente através do sistema nacional de notificação listado no Apêndice V. efeitos secundários pode ajudar fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
O Remicade geralmente é armazenado por profissionais de saúde. Se você precisar, os detalhes de retenção são os seguintes:
- Mantenha este medicamento fora da vista e do alcance das crianças.
- Não utilize este medicamento após o prazo de validade impresso no rótulo e na embalagem exterior a seguir a “VAL.” O prazo de validade corresponde ao último dia desse mês.
- Conservar no frigorífico (2 ° C - 8 ° C).
- Este medicamento também pode ser conservado na embalagem original, fora do frigorífico, até um máximo de 25 ° C por um único período de até seis meses. Nessa situação, não deve ser armazenado novamente na geladeira. Escreva a nova data de validade na caixa, incluindo dia / mês / ano. Descarte este medicamento se não for usado até o novo prazo de validade ou até o prazo de validade impresso na embalagem, o que ocorrer primeiro.
- Quando Remicade é preparado para perfusão, recomenda-se que seja utilizado o mais cedo possível (dentro de 3 horas). No entanto, se a solução for preparada em condições totalmente isentas de germes, pode ser conservada no frigorífico durante 24 horas entre 2 ° C e 8 ° C
- Não use este medicamento se estiver descolorido ou contiver partículas.
O que Remicade contém
- O ingrediente ativo é o infliximabe. Cada frasco para injetáveis contém 100 mg de infliximab. Após a preparação, cada ml contém 10 mg de infliximab.
- Os outros componentes são sacarose, polissorbato 80, fosfato de sódio monobásico e fosfato de sódio dibásico.
Qual a aparência de Remicade e conteúdo da embalagem
Remicade é fornecido em frascos para injectáveis de vidro contendo o pó para concentrado para solução para perfusão. O pó consiste em grânulos brancos liofilizados.
Remicade está disponível em embalagens de 1, 2, 3, 4 ou 5 frascos para injectáveis. Nem todos os tamanhos de embalagem podem ser comercializados
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
REMICADE 100 MG EM PÓ PARA CONCENTRADO PARA SOLUÇÃO PARA INFUSÃO
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Cada frasco para injetáveis contém 100 mg de infliximab. O infliximabe é um anticorpo monoclonal IgG1 humano-murino quimérico produzido em células de hibridoma murino por tecnologia de DNA recombinante. Após reconstituição, cada ml contém 10 mg de infliximab.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Pó para concentrado para solução para perfusão.
O pó consiste em grânulos brancos liofilizados.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
Artrite reumatóide
Remicade, em combinação com metotrexato, é indicado para a redução dos sinais e sintomas e a melhora da função física em:
• pacientes adultos com doença ativa quando a resposta aos medicamentos antirreumáticos modificadores da doença (DMARDs), incluindo metotrexato, foi inadequada.
• pacientes adultos com doença grave, ativa e progressiva não previamente tratados com metotrexato ou outros DMARDs.
Uma redução na taxa de progressão das lesões articulares foi demonstrada por avaliação radiográfica nesta população de doentes (ver secção 5.1).
Doença de Crohn em adultos
Remicade é indicado para:
• o tratamento da doença de Crohn ativa moderada a grave em pacientes adultos que não responderam, apesar do tratamento completo e adequado com corticosteroides e / ou imunossupressores; ou em pacientes que não toleram ou têm contra-indicações médicas para as terapias mencionadas.
• o tratamento da doença de Crohn fistulizante ativa em pacientes adultos que não responderam, apesar de um curso de terapia completo e adequado com o tratamento convencional (incluindo antibióticos, drenagem e terapia imunossupressora).
Doença de Crohn em crianças
Remicade é indicado para o tratamento da doença de Crohn ativa grave, em crianças e adolescentes de 6 a 17 anos que não responderam à terapia convencional com um corticosteroide, um imunomodulador e terapia nutricional primária, ou em pacientes que não toleram ou têm contra-indicações para as terapias acima mencionadas. Remicade só foi estudado em combinação com terapia imunossupressora convencional.
Colite ulcerativa
Remicade é indicado para o tratamento de colite ulcerativa ativa moderada a grave em pacientes adultos que não responderam adequadamente à terapia convencional, incluindo corticosteroides e 6-mercaptopurina (6-MP) ou azatioprina (AZA), ou que são intolerantes ou para os quais há uma contra-indicação médica para essas terapias.
Colite ulcerosa pediátrica
Remicade é indicado para o tratamento da colite ulcerosa ativa grave em crianças e adolescentes de 6 a 17 anos de idade que não responderam adequadamente à terapia convencional, incluindo corticosteroides e 6-MP ou AZA, ou que têm intolerância ou para os quais haja um médico contra-indicação a essas terapias.
Espondilite anquilosante
Remicade é indicado para o tratamento da espondilite anquilosante ativa grave em pacientes adultos que não responderam adequadamente às terapias convencionais.
Artrite psoriática
Remicade é indicado para o tratamento da artrite psoriática ativa e progressiva em pacientes adultos, quando a resposta aos tratamentos DMARD anteriores foi inadequada.
Remicade deve ser administrado:
• em associação com metotrexato
• ou individualmente em pacientes que são intolerantes ao metotrexato ou para os quais ele é contra-indicado
Remicade demonstrou melhorar a função física em doentes com artrite psoriática e reduzir a taxa de progressão da lesão articular periférica medida por raios-X em doentes com subtipos poliarticulares simétricos da doença (ver secção 5.1).
Psoríase
Remicade está indicado para o tratamento da psoríase em placas moderada a grave em doentes adultos que falharam ou estão contra-indicados ou que foram intolerantes a outros tratamentos sistémicos incluindo ciclosporina, metotrexato ou PUVA (ver secção 5.1).
04.2 Posologia e método de administração
O tratamento com Remicade deve ser iniciado e supervisionado por médicos especialistas com experiência no diagnóstico e tratamento da artrite reumatóide, doença inflamatória do intestino, espondilite anquilosante, artrite psoriática ou psoríase. Remicade deve ser administrado por via intravenosa. As perfusões de Remicade devem ser administradas por profissionais de saúde qualificados, com formação no reconhecimento de quaisquer questões relacionadas com a perfusão.Os doentes tratados com Remicade devem receber o folheto informativo e o Cartão de Alerta do Doente.
Durante o tratamento com Remicade, o uso de outras terapias concomitantes, como corticosteróides e imunossupressores, deve ser otimizado.
Dosagem
Adultos (≥ 18 anos)
Artrite reumatóide
Uma infusão intravenosa de 3 mg / kg seguida por infusões adicionais de 3 mg / kg nas semanas 2 e 6 após a primeira infusão e, em seguida, a cada 8 semanas.
Remicade deve ser administrado concomitantemente com metotrexato.
Os dados disponíveis sugerem que a resposta clínica é geralmente alcançada dentro de 12 semanas após o início do tratamento. Se um paciente tiver uma resposta inadequada ou perder a resposta após esse período, um aumento gradual na dosagem de 1,5 mg pode ser considerado. / Kg, até um máximo de 7,5 mg / kg, a cada 8 semanas. Alternativamente, pode ser considerada a administração de 3 mg / kg a cada 4 semanas. Se for alcançada uma resposta adequada, o tratamento deve continuar. pacientes com a dosagem ou frequência escolhida.Deve-se considerar cuidadosamente a continuação da terapia em pacientes que não mostram evidência de benefício terapêutico nas primeiras 12 semanas de tratamento ou após o ajuste da dose.
Doença de Crohn ativa moderada a grave
5 mg / kg administrados como uma infusão intravenosa seguida por uma infusão adicional de 5 mg / kg 2 semanas após a primeira infusão. Se um paciente não responder à terapia após 2 doses, nenhum tratamento adicional com infliximabe deve ser administrado. Os dados disponíveis não apoiam o tratamento adicional com infliximabe em não pacientes respondentes dentro de 6 semanas após a primeira infusão.
Em pacientes que respondem, as soluções alternativas para o tratamento continuado são:
• Manutenção: infusão suplementar de 5 mg / kg na semana 6 após a primeira dose, seguida por infusões repetidas a cada 8 semanas ou
• Re-administração: uma perfusão de 5 mg / kg se os sinais e sintomas da doença persistirem (ver “Re-administração” e secção 4.4).
Embora faltem dados comparativos, os dados limitados em doentes que responderam inicialmente à terapêutica de 5 mg / kg mas perderam a resposta indicam que alguns doentes podem recuperar a resposta aumentando a dose (ver secção 5.1). A continuação da terapia deve ser cuidadosamente reconsiderada em pacientes que não mostram evidência de benefício terapêutico após o ajuste da dose.
Doença de Crohn fistulizante ativa
5 mg / kg administrados como uma infusão intravenosa seguida por infusões adicionais de 5 mg / kg na 2ª e 6ª semanas após a primeira infusão. Se um paciente não responder após 3 doses, nenhum tratamento adicional com infliximabe deve ser administrado.
Em pacientes que respondem, as soluções alternativas para o tratamento continuado são:
• Manutenção: infusões adicionais de 5 mg / kg a cada 8 semanas ou
• Re-administração: uma infusão de 5 mg / kg se os sinais e sintomas da doença persistirem, seguida por infusões de 5 mg / kg a cada 8 semanas (ver “Re-administração” e secção 4.4).
Embora faltem dados comparativos, os dados limitados em doentes que responderam inicialmente à terapêutica de 5 mg / kg mas perderam a resposta indicam que alguns doentes podem recuperar a resposta aumentando a dose (ver secção 5.1). A continuação da terapia deve ser cuidadosamente reconsiderada em pacientes que não mostram evidência de benefício terapêutico após o ajuste da dose.
Na doença de Crohn, a experiência de re-administração, se os sinais e sintomas da doença persistirem, é limitada e não há dados comparativos de risco / benefício de soluções alternativas para o tratamento continuado.
Colite ulcerativa
Uma infusão intravenosa de 5 mg / kg seguida por infusões adicionais de 5 mg / kg nas semanas 2 e 6 após a primeira infusão, então repetida a cada 8 semanas.
Os dados disponíveis sugerem que a resposta clínica é geralmente alcançada dentro de 14 semanas após o início do tratamento, ou seja, após três administrações. Deve-se considerar cuidadosamente a continuação da terapia em pacientes que não respondem dentro deste período de tempo.
Espondilite anquilosante
Uma infusão intravenosa de 5 mg / kg seguida por infusões adicionais de 5 mg / kg nas semanas 2 e 6 após a primeira infusão, e então repetida após 6 a 8 semanas. Se um paciente não responder em 6 semanas (ou seja, após 2 doses), ele não deve receber nenhum tratamento adicional com infliximabe.
Artrite psoriática
Uma infusão intravenosa de 5 mg / kg seguida por infusões adicionais de 5 mg / kg nas semanas 2 e 6 após a primeira infusão, então repetida a cada 8 semanas.
Psoríase
Uma infusão intravenosa de 5 mg / kg seguida por infusões adicionais de 5 mg / kg nas semanas 2 e 6 após a primeira infusão, então repetida a cada 8 semanas. Se um paciente não responder em 14 semanas (ou seja, após 4 doses), nenhum tratamento adicional com infliximabe deve ser administrado.
Re-administração para doença de Crohn e artrite reumatóide
Se os sinais e sintomas da doença recorrerem, Remicade pode ser administrado novamente dentro de 16 semanas após a última perfusão. Em estudos clínicos, as reações de hipersensibilidade retardada foram "pouco frequentes" e ocorreram após intervalos sem administração de Remicade. Menos de 1 ano (ver secções 4.4 e 4.8) A segurança e eficácia da readministração não foram estabelecidas após mais de 16 semanas sem administração de Remicade. Isso se aplica a pacientes com doença de Crohn e pacientes com artrite reumatóide.
Re-administração para colite ulcerosa
A segurança e eficácia de readministrações em intervalos diferentes de 8 semanas não foram estabelecidas (ver secções 4.4 e 4.8).
Re-administração para espondilite anquilosante
A segurança e eficácia de readministrações além das administradas com um intervalo de 6 a 8 semanas não foram estabelecidas (ver secções 4.4 e 4.8).
Re-administração para artrite psoriática
A segurança e eficácia de readministrações em intervalos diferentes de 8 semanas não foram estabelecidas (ver secções 4.4 e 4.8).
Re-administração para psoríase
Uma "experiência limitada na psoríase resultante do retratamento com uma única dose de Remicade após um intervalo de 20 semanas sugere uma" eficácia reduzida e uma "maior incidência de reações à perfusão ligeiras a moderadas" quando comparada com o regime de indução inicial. (Ver seção 5.1).
Uma “experiência limitada com retratamento após agravamento da doença através de um regime de re-indução sugere uma“ elevada incidência de reações à perfusão, incluindo as graves, quando comparadas com as de 8 semanas de tratamento de manutenção (ver parágrafo 4.8).
Re-administração nas diferentes indicações
No caso de a terapia de manutenção ser descontinuada e houver necessidade de reiniciar o tratamento, o uso de um regime de re-indução não é recomendado (ver seção 4.8) .Nesta situação, o tratamento com Remicade deve ser reiniciado em dose única seguida de dose de manutenção de acordo com as recomendações descritas acima.
Pacientes idosos (≥ 65 anos)
Não foram realizados estudos específicos com Remicade em pacientes idosos. Não foram observadas diferenças substanciais relacionadas à idade na depuração ou volume de distribuição em estudos clínicos.
Não é necessário ajuste de dose (ver seção 5.2). Para mais informações sobre a segurança de Remicade em doentes idosos, ver secções 4.4 e 4.8.
Função renal e / ou hepática prejudicada
Remicade não foi estudado nestas populações de pacientes. Nenhuma recomendação de dose pode ser feita (ver seção 5.2).
População pediátrica
Doença de Crohn (6 - 17 anos)
Uma dose de 5 mg / kg administrada por infusão intravenosa seguida por infusões subsequentes de doses de 5 mg / kg 2 e 6 semanas após a primeira infusão e a cada 8 semanas a partir de então. Os dados disponíveis não apoiam o tratamento adicional com infliximab em crianças e adolescentes que não respondem nas primeiras 10 semanas de tratamento (ver secção 5.1).
Alguns pacientes podem requerer um intervalo de dose mais curto para manter o benefício clínico, enquanto para outros um intervalo de dose mais longo pode ser suficiente. Os pacientes que tiveram o intervalo de tempo entre as doses reduzido para menos de 8 semanas podem apresentar risco aumentado de reações adversas. A continuação da terapia com um intervalo mais curto deve ser cuidadosamente considerada em pacientes que não mostram evidência de benefício terapêutico. Após uma alteração no intervalo de tempo entre as doses.
A segurança e eficácia de Remicade em crianças com doença de Crohn com menos de 6 anos de idade não foram estudadas. Os dados farmacocinéticos atualmente disponíveis são descritos na seção 5.2, mas nenhuma recomendação posológica pode ser feita em crianças com menos de 6 anos de idade.
Colite ulcerativa (6 - 17 anos)
Uma dose de 5 mg / kg administrada por infusão intravenosa seguida por infusões subsequentes de doses de 5 mg / kg 2 e 6 semanas após a primeira infusão e a cada 8 semanas a partir de então. Os dados disponíveis não apoiam o tratamento adicional com infliximab em doentes pediátricos que não respondem nas primeiras 8 semanas de tratamento (ver secção 5.1).
A segurança e eficácia de Remicade em crianças com colite ulcerosa com menos de 6 anos de idade não foram estudadas. Os dados farmacocinéticos atualmente disponíveis são descritos na seção 5.2, mas nenhuma recomendação posológica pode ser feita em crianças com menos de 6 anos de idade.
Psoríase
A segurança e eficácia de Remicade em crianças e adolescentes com menos de 18 anos de idade na indicação psoríase não foram estabelecidas Os dados atualmente disponíveis estão descritos na secção 5.2, mas não pode ser feita qualquer recomendação posológica.
Artrite idiopática juvenil, artrite psoriática e espondilite anquilosante
A segurança e eficácia de Remicade em crianças e adolescentes com menos de 18 anos de idade nas indicações artrite idiopática juvenil, artrite psoriática e espondilite anquilosante não foram estabelecidas. Os dados atualmente disponíveis são descritos na seção 5.2, mas nenhuma recomendação posológica pode ser feita.
Artrite reumatóide juvenil
A segurança e eficácia de Remicade em crianças e adolescentes com menos de 18 anos de idade na indicação para artrite reumatóide juvenil não foram estabelecidas. Os dados atualmente disponíveis estão descritos nas secções 4.8 e 5.2, mas não pode ser feito. Recomendação sobre a posologia.
Função renal e / ou hepática prejudicada
Remicade não foi estudado nestas populações de pacientes. Nenhuma recomendação de dose pode ser feita (ver seção 5.2).
Método de administração
Remicade deve ser administrado por via intravenosa durante um período de 2 horas. Todos os pacientes tratados com Remicade devem ser observados por pelo menos 1-2 horas após a infusão para reações agudas relacionadas à infusão. Equipamentos de emergência, como adrenalina, anti-histamínicos, corticosteroides e um respirador artificial devem ser mantidos disponíveis. Os pacientes podem ser pré-tratados com, por exemplo, um anti-histamínico, hidrocortisona e / ou paracetamol e a taxa de infusão pode ser diminuída para reduzir o risco relacionado à infusão reações, especialmente se reações relacionadas com a perfusão ocorreram anteriormente (ver secção 4.4).
Infusões abreviadas em indicações para adultos
Em pacientes adultos cuidadosamente selecionados que toleraram pelo menos 3 infusões iniciais de Remicade de 2 horas (fase de indução) e que estão recebendo terapia de manutenção, administração de infusões subsequentes por um período não inferior a 1 hora Se uma reação à infusão associada à infusão reduzida ocorrer, uma taxa de infusão mais lenta pode ser considerada para infusões futuras, caso o tratamento continue. Não foram estudadas perfusões abreviadas em doses> 6 mg / kg (ver secção 4.8).
Para obter instruções sobre a preparação e administração, consulte a seção 6.6.
04.3 Contra-indicações
Doentes com história de hipersensibilidade ao infliximab (ver secção 4.8), a outras proteínas murinas ou a qualquer um dos excipientes listados na secção 6.1.
Doentes com tuberculose ou outras infecções graves, como sépsis, abcessos e infecções oportunistas (ver secção 4.4).
Doentes com insuficiência cardíaca moderada a grave (NYHA - New York Heart Association - Classe III / IV) (ver secções 4.4 e 4.8).
04.4 Advertências especiais e precauções adequadas de uso
Para melhorar a rastreabilidade dos medicamentos biológicos, a marca comercial e o número do lote do medicamento administrado devem ser claramente registados (ou marcados) no registo do doente.
Reações à infusão e hipersensibilidade
O infliximab foi associado a reações agudas relacionadas com a perfusão, incluindo choque anafilático e reações de hipersensibilidade retardada (ver secção 4.8).
Podem ocorrer reações agudas à perfusão, incluindo reações anafiláticas, durante (segundos) ou horas após a perfusão. Se ocorrerem reações agudas à infusão, a infusão deve ser interrompida imediatamente. Equipamentos de emergência, como adrenalina, anti-histamínicos, corticosteroides e um ventilador artificial devem ser mantidos disponíveis.Os pacientes podem ser pré-tratados com, por exemplo, um anti-histamínico, hidrocortisona e / ou paracetamol para prevenir efeitos leves e transitórios.
Podem desenvolver-se anticorpos ao infliximab e têm sido associados a um aumento da frequência de reações à perfusão. Uma baixa taxa de reações à infusão foram reações alérgicas graves.Também foi observada uma associação entre o desenvolvimento de anticorpos ao infliximabe e uma redução na resposta. A administração concomitante de imunomoduladores foi associada a uma menor incidência de anticorpos ao infliximabe e uma redução na frequência das reações à infusão.O efeito da terapia imunomoduladora concomitante foi mais intenso em pacientes tratados episodicamente do que em pacientes recebendo terapia de manutenção. Os doentes que descontinuaram a terapêutica imunossupressora antes ou durante o tratamento com Remicade apresentam um risco aumentado de desenvolver estes anticorpos. Os anticorpos ao infliximabe nem sempre podem ser detectados em amostras de soro. Se ocorrerem reações graves, deve ser administrado tratamento sintomático e não devem ser administradas perfusões adicionais de Remicade (ver secção 4.8).
Reações de hipersensibilidade tardia foram relatadas em estudos clínicos. Os dados disponíveis sugerem um risco aumentado de hipersensibilidade retardada ao aumento da duração dos intervalos de tempo sem administração de Remicade. Os doentes devem ser aconselhados a contactar o seu médico imediatamente no caso de um acontecimento adverso retardado (ver secção 4.8). Se os doentes voltarem a ser tratados após um período prolongado período, eles devem ser monitorados de perto quanto a sinais e sintomas de hipersensibilidade retardada.
Infecções
Os doentes devem ser cuidadosamente monitorizados quanto a infecções, incluindo tuberculose, antes, durante e após o tratamento com Remicade. Uma vez que a eliminação do infliximab pode demorar até seis meses, a monitorização deve continuar durante este período.O tratamento adicional com Remicade não deve ser administrado se o doente desenvolver infecções graves ou sépsis.
É necessário cuidado ao usar Remicade em pacientes com infecção crônica ou história de infecções recorrentes, incluindo terapia concomitante com imunossupressores.Os pacientes devem ser devidamente alertados sobre a necessidade de evitar a exposição a fatores de risco potenciais para infecções.
O fator de necrose tumoral alfa (TNFα) medeia a inflamação e modula as respostas imunes celulares. Os dados experimentais demonstram que o TNFα é essencial para a resolução de infecções intracelulares. A experiência clínica mostra que as defesas imunológicas do hospedeiro estão comprometidas em alguns pacientes tratados com infliximabe.
Deve-se notar que a supressão do TNFα pode mascarar os sintomas de uma infecção, como febre. O reconhecimento precoce de manifestações clínicas atípicas de infecções graves e manifestações clínicas típicas de infecções raras e incomuns é fundamental para minimizar atrasos no diagnóstico e tratamento.
Os pacientes que tomam drogas bloqueadoras de TNF são mais propensos a infecções graves.
Tuberculose, infecções bacterianas, incluindo sepse e pneumonia, infecções invasivas fúngicas, virais e outras infecções oportunistas foram observadas em pacientes tratados com infliximabe. Algumas dessas infecções foram fatais; as infecções oportunistas mais frequentemente relatadas com uma taxa de mortalidade> 5% incluem penumocistose, candidíase, listeriose e aspergilose.
Os doentes que desenvolvem uma nova infecção durante o tratamento com Remicade devem ser cuidadosamente monitorizados e submetidos a uma avaliação diagnóstica completa. A administração de Remicade deve ser interrompida se um paciente desenvolver uma nova infecção grave ou sepse e iniciar uma terapia antimicrobiana ou antifúngica apropriada até que a infecção seja resolvida.
Tuberculose
Foram notificados casos de tuberculose ativa em doentes tratados com Remicade. Ressalta-se que, na maioria dos casos, tratava-se de tuberculose extrapulmonar, tanto localizada quanto difusa.
Antes de iniciar o tratamento com Remicade, todos os pacientes devem ser avaliados para tuberculose ativa e inativa ("latente"). Esta avaliação deve incluir uma história médica detalhada, incluindo uma história pessoal de tuberculose ou possível contato anterior com uma fonte de infecção de TB e terapias imunossupressoras anteriores e / ou concomitantes. Devem ser realizados testes diagnósticos adequados, como testes cutâneos de tuberculina e radiografia de tórax em todos os pacientes (podem ser aplicadas as diretrizes locais). Recomenda-se que esses testes sejam relatados no Cartão de Alerta do Paciente.Os prescritores são lembrados do risco de resultados falso-negativos do teste tuberculínico, particularmente em pacientes gravemente enfermos ou imunocomprometidos.
Se for diagnosticada tuberculose ativa, a terapia com Remicade não deve ser iniciada. (ver seção 4.3)
Se houver suspeita de tuberculose latente, um médico com experiência no tratamento da tuberculose deve ser consultado. Em todas as situações descritas abaixo, a relação benefício / risco da terapêutica com Remicade deve ser cuidadosamente considerada.
Se a tuberculose inativa ("latente") for diagnosticada, a terapia anti-tuberculose para a tuberculose latente deve ser iniciada antes de iniciar a terapia com Remicade, de acordo com as diretrizes locais.
Em pacientes com muitos fatores de risco ou fatores de risco significativos para tuberculose e com teste negativo para tuberculose latente, a terapia antituberculose deve ser considerada antes do início de Remicade.
A utilização de terapêutica antituberculose também deve ser considerada antes do início da terapêutica com Remicade em doentes com história prévia de tuberculose latente ou ativa, para os quais um curso de tratamento adequado não pode ser confirmado.
Alguns casos de tuberculose ativa foram relatados em pacientes tratados com Remicade durante e após o tratamento da tuberculose latente.
Todos os doentes devem ser aconselhados a procurar aconselhamento médico se surgirem sinais / sintomas sugestivos de tuberculose (por exemplo, tosse persistente, emagrecimento / perda de peso, febre baixa) durante ou após o tratamento com Remicade.
Infecções fúngicas invasivas
Uma infecção fúngica invasiva, como aspergilose, candidíase, pneumocistose, histoplasmose, coccidioidomicose ou blastomicose, deve ser suspeitada em pacientes tratados com Remicade se desenvolverem doença sistêmica grave e um médico competente no diagnóstico e tratamento de infecções fúngicas invasivas deve ser consultado em estágio inicial ao visitar esses pacientes. As infecções fúngicas invasivas podem se apresentar como doença disseminada em vez de localizada, e os testes de antígeno e anticorpos podem ser negativos em alguns pacientes com infecção ativa. A terapia antifúngica empírica apropriada deve ser considerada no processo de diagnóstico, levando em consideração o risco de uma infecção fúngica grave e os riscos da terapia antifúngica.
Para pacientes que viveram ou viajaram para regiões onde infecções fúngicas invasivas, como histoplasmose, coccidioidomicose ou blastomicose, são endêmicas, os benefícios e riscos do tratamento com Remicade devem ser cuidadosamente considerados antes de iniciar a terapia com Remicade.
Doença de Crohn Fistulizante
Os doentes com doença de Crohn fistulizante com fístulas supurativas agudas não devem iniciar a terapêutica com Remicade até que uma possível fonte de infecção, particularmente abcessos, tenha sido excluída (ver secção 4.3).
Reativação da hepatite B (HBV)
A reativação da hepatite B foi observada em pacientes tratados com um antagonista do TNF, incluindo infliximabe e que eram portadores crônicos deste vírus. Em alguns casos, ocorreram desfechos fatais.
Os pacientes devem ser avaliados para infecção pelo VHB antes de iniciar o tratamento com Remicade.Para os pacientes com teste positivo para infecção pelo VHB, é recomendada a consulta com um médico com experiência no tratamento da hepatite B.
Os portadores do VHB que requerem tratamento com Remicade devem ser monitorados de perto quanto a sinais e sintomas de infecção VHB ativa ao longo da terapia e por muitos meses após o final da terapia. Dados insuficientes estão disponíveis em pacientes com VHB. Tratados com terapia antiviral em combinação com antagonista do TNF terapêutica para prevenir a reativação do VHB Em doentes que desenvolvam a reativação do VHB, o tratamento com Remicade deve ser descontinuado e deve ser iniciada uma terapêutica antiviral eficaz com tratamento de suporte adequado.
Eventos hepatobiliares
Durante o período de comercialização de Remicade, foram observados casos muito raros de icterícia e hepatite não infecciosa, alguns com características de hepatite autoimune. Houve casos isolados de insuficiência hepática resultando em transplante de fígado ou morte. Em pacientes com sinais e sintomas de disfunção hepática, o nível de lesão hepática deve ser avaliado. Se ocorrer icterícia e / ou elevações de ALT ≥ 5 vezes o limite superior do normal, o tratamento com Remicade deve ser interrompido e deve ser realizado um exame completo das condições anormais.
Associação de um inibidor de TNF-alfa e anakinra
Infecções graves e neutropenia ocorreram em ensaios clínicos combinados de anakinra e outro inibidor do TNFα, sem nenhum benefício clínico adicional sobre o uso de etanercepte sozinho. Dada a natureza dos eventos adversos observados com a combinação de etanercepte e anakinra, toxicidades semelhantes podem ocorrer com o combinação de anakinra e outros inibidores do TNFα Portanto, a combinação de Remicade e anakinra não é recomendada.
Associação de um inibidor de TNF-alfa e abatacepte
Em ensaios clínicos, a utilização combinada de antagonistas do TNF e abatacept foi associada a um risco aumentado de infecções, incluindo infecções graves, em comparação com os antagonistas do TNF utilizados isoladamente, sem um aumento do benefício clínico. Remicade e abatacept não são recomendados.
Associação com outras terapias biológicas
Não há informações suficientes sobre o uso concomitante de infliximabe com outras terapias biológicas usadas para tratar as mesmas condições que o infliximabe. O uso concomitante de infliximabe com esses produtos biológicos não é recomendado devido à possibilidade de um risco aumentado de infecção e outras potenciais interações medicamentosas.
Substituição entre DMARDs biológicos
Deve-se ter cuidado e os pacientes devem continuar a ser monitorados ao mudar de um produto biológico para outro, pois a atividade biológica sobreposta pode aumentar ainda mais o risco de eventos adversos, incluindo infecção.
Vacinas vivas / agentes terapêuticos infecciosos
Em pacientes tratados com terapia anti-TNF, dados limitados estão disponíveis sobre a resposta à vacinação com vacinas vivas ou sobre a transmissão secundária da infecção com a administração de vacinas vivas.O uso de vacinas vivas pode levar a infecções clínicas, incluindo infecções disseminadas. . A co-administração de vacinas vivas com Remicade não é recomendada.
Em crianças expostas in utero ao infliximabe, foi relatado um desfecho fatal devido à infecção disseminada pelo bacilo de Calmette-Guérin (BCG) após a administração da vacina BCG após o nascimento. Antes de administrar vacinas vivas a bebês expostos no utero um período de espera de pelo menos seis meses após o nascimento é recomendado para o infliximabe (ver seção 4.6).
Outros usos de agentes terapêuticos infecciosos, como bactérias vivas atenuadas (por exemplo, instilações intravesicais com BCG para tratamento de câncer) podem resultar em infecções clínicas, incluindo infecções disseminadas. Recomenda-se que os agentes infecciosos terapêuticos não sejam administrados concomitantemente com Remicade.
Reações autoimunes
A deficiência relativa de TNFα causada pela terapia anti-TNF pode levar ao início de um processo autoimune. Se um paciente tiver sintomas preditivos de uma síndrome semelhante ao lúpus após o tratamento com Remicade e for positivo para anticorpos anti-DNA para dupla hélice, mais o tratamento com Remicade não deve ser administrado (ver secção 4.8).
Efeitos no sistema nervoso
O uso de agentes bloqueadores de TNF, incluindo infliximabe, foi associado ao início ou exacerbação de sintomas clínicos e / ou evidências radiográficas de distúrbios desmielinizantes do sistema nervoso central, incluindo esclerose múltipla e distúrbios desmielinizantes periféricos, incluindo síndrome de Guillain-Barré em pacientes com doenças desmielinizantes pré-existentes ou recentes, os benefícios e riscos do tratamento com anti-TNF devem ser cuidadosamente considerados antes de iniciar a terapia com Remicade.
A descontinuação da terapia com Remicade deve ser considerada se essas condições se desenvolverem.
Neoplasias malignas e doenças linfoproliferativas
Mais casos de doenças malignas, incluindo linfoma, foram observados em fases controladas de ensaios clínicos com inibidores de TNF entre pacientes que receberam um inibidor de TNF em comparação com pacientes de controle. Durante os ensaios clínicos com Remicade, em todas as indicações aprovadas, a incidência de linfoma em doentes tratados com Remicade foi superior ao esperado na população em geral, mas a frequência de linfoma foi rara. Na experiência pós-comercialização, foram notificados casos de leucemia em pacientes tratados com um antagonista do TNF. Há um risco aumentado de desenvolvimento de linfoma e leucemia em pacientes com artrite reumatóide com uma doença inflamatória muito ativa e de longa data que complica a avaliação de risco.
Em um ensaio clínico exploratório que avaliou o uso de Remicade em pacientes com doença pulmonar obstrutiva crônica moderada a grave (Doença de obstrução pulmonar crônica, DPOC), foram notificados mais casos de doenças malignas em doentes tratados com Remicade do que em doentes de controlo. Todos os pacientes eram fumantes inveterados. Deve-se ter cuidado ao avaliar o tratamento de pacientes com risco aumentado de malignidade como fumantes inveterados.
Com base no conhecimento atual, não pode ser excluído o risco de desenvolver linfomas ou doenças malignas em doentes tratados com um inibidor do TNF (ver secção 4.8). Deve-se ter cuidado ao considerar a terapia com inibidores de TNF em pacientes com história de malignidade ou ao considerar o tratamento prolongado em pacientes que desenvolvem malignidade.
Deve-se ter cautela também em pacientes com psoríase que já foram tratados extensivamente com imunossupressores ou por períodos prolongados com PUVA.
Na experiência pós-comercialização, foram notificados casos de neoplasias, algumas das quais fatais, entre crianças, adolescentes e adultos jovens (até 22 anos) tratados com fármacos bloqueadores do TNF (início da terapêutica ≤ 18 anos de idade), incluindo Remicade. metade dos casos eram linfomas.Os outros casos eram representados por uma variedade de neoplasias diferentes e incluíam neoplasias raras geralmente associadas à imunossupressão. Não pode ser excluído o risco de desenvolvimento de doenças malignas em pacientes tratados com inibidores do TNF.
Foram notificados casos raros pós-comercialização de linfoma hepatoesplénico das células T (HSTCL) em doentes tratados com agentes bloqueadores do TNF, incluindo infliximab. Esta forma rara de linfoma das células T tem um curso e resultado extremamente agressivos. Geralmente fatal. Quase todos os doentes tinham recebido tratamento com AZA ou 6-MP simultaneamente ou imediatamente antes de um bloqueador de TNF. A grande maioria dos casos com Remicade ocorreu em doentes com doença de Crohn ou colite ulcerosa e A maioria dos casos foi notificada em adolescentes ou adultos jovens do sexo masculino. o risco potencial da combinação de AZA ou 6-MP e Remicade deve ser cuidadosamente considerado. Não pode ser excluído o risco de desenvolvimento de linfoma hepatoesplénico das células T em doentes tratados com Remicade (ver secção 4.8).
Foram notificados melanoma e carcinoma de células de Merkel em doentes a receber terapêutica com um bloqueador do TNF, incluindo Remicade (ver secção 4.8). O exame periódico da pele é recomendado, principalmente para pacientes com fatores de risco para câncer de pele.
Um estudo de coorte retrospectivo baseado em dados de registros nacionais de saúde suecos encontrou um aumento na incidência de câncer cervical em mulheres com artrite reumatóide tratadas com infliximabe em comparação com pacientes não tratadas biologicamente ou na população em geral, incluindo aquelas com mais de 60 anos de idade. O rastreamento periódico deve continuar em mulheres tratados com Remicade, incluindo os maiores de 60 anos.
Todos os pacientes com colite ulcerosa que têm um risco aumentado de desenvolver displasia do cólon ou carcinoma (por exemplo, pacientes com colite ulcerosa de longa data ou colangite esclerosante primária) ou que têm um histórico médico de displasia ou câncer de cólon devem ser investigados. Em relação a esta displasia em intervalos regulares, antes do início da terapia e durante o curso da doença. Essa avaliação deve incluir uma colonoscopia e biópsias de acordo com as diretrizes locais. À luz dos dados atuais, não se sabe se o tratamento com infliximab afeta o risco de desenvolver displasia ou cancro do cólon (ver secção 4.8).
Uma vez que não foi estabelecida a possibilidade de um risco aumentado de desenvolver cancro em doentes tratados com Remicade com displasia diagnosticada recentemente, é necessário avaliar a relação benefício / risco nos doentes individuais e considerar a interrupção da terapêutica.
Insuficiência cardíaca
Remicade deve ser usado com cautela em pacientes com insuficiência cardíaca leve (classe I / II da NYHA). Os doentes devem ser monitorizados de perto e o tratamento com Remicade deve ser descontinuado em doentes que apresentem sintomas novos ou agravamento de insuficiência cardíaca (ver secções 4.3 e 4.8).
Reações hematológicas
Foram notificados casos de pancitopenia, leucopenia, neutropenia e trombocitopenia em doentes a receber medicamentos anti-TNF, incluindo Remicade. Todos os pacientes devem ser aconselhados a procurar atendimento médico imediato se desenvolverem sinais ou sintomas compatíveis de discrasias sanguíneas (por exemplo, febre persistente, hematomas, sangramento e palidez). A descontinuação da terapêutica com Remicade deve ser considerada em doentes com anomalias hematológicas significativas confirmadas.
Outros
A experiência com a segurança do tratamento com Remicade em pacientes que foram submetidos a cirurgia, incluindo artroplastia, é limitada. A longa meia-vida de eliminação do infliximabe deve ser considerada no planejamento da cirurgia. Um doente que necessite de cirurgia durante o tratamento com Remicade deve ser cuidadosamente monitorizado para deteção de um risco aumentado de infecções e devem ser consideradas as medidas adequadas.
A falta de resposta ao tratamento para a doença de Crohn pode indicar a presença de estenoses fibróticas rígidas que podem exigir tratamento cirúrgico. Não há evidências clínicas que sugiram que o infliximabe piora ou causa estenoses fibróticas.
Populações especiais
Pacientes idosos (≥ 65 anos)
A incidência de infecções graves em doentes com idade igual ou superior a 65 anos tratados com Remicade foi superior à de doentes com idade inferior a 65 anos. Algumas destas infecções foram fatais. Deve ser dada especial atenção ao risco de infecção no tratamento de idosos (ver secção 4.8) .
População pediátrica
Infecções
Em estudos clínicos, as infecções foram notificadas com mais frequência em populações pediátricas do que em populações adultas (ver secção 4.8).
Vacinas
Recomenda-se que os pacientes pediátricos, se possível, recebam todas as vacinas de acordo com as diretrizes mais recentes antes de iniciar a terapia com Remicade.
Neoplasias malignas e doenças linfoproliferativas
Na experiência pós-comercialização, foram notificados casos de neoplasias, algumas das quais fatais, entre crianças, adolescentes e adultos jovens (até 22 anos) tratados com fármacos bloqueadores do TNF (início da terapêutica ≤ 18 anos de idade), incluindo Remicade. metade dos casos eram linfomas.Os outros casos eram representados por uma variedade de neoplasias diferentes e incluíam neoplasias raras geralmente associadas à imunossupressão. Não pode ser excluído o risco de desenvolvimento de neoplasias malignas em crianças e adolescentes tratados com inibidores do TNF.
Casos raros de linfoma hepatoesplênico de células T foram relatados após a comercialização em pacientes tratados com agentes bloqueadores de TNF, incluindo infliximabe. Esta forma rara de linfoma de células T tem um curso extremamente agressivo e geralmente fatal. Quase todos os pacientes receberam tratamento. com AZA ou 6-MP simultaneamente ou imediatamente antes de um bloqueador de TNF. A grande maioria dos casos com Remicade ocorreu em pacientes com doença de Crohn ou colite ulcerosa e a maioria dos casos foi relatada em adolescentes ou adultos jovens do sexo masculino. combinação de AZA ou 6-MP e Remicade deve ser cuidadosamente considerada. Não pode ser excluído o risco de desenvolvimento de linfoma hepatoesplénico das células T em doentes tratados com Remicade (ver secção 4.8).
04.5 Interações com outros medicamentos e outras formas de interação
Não foram realizados estudos de interação.
Existem indicações de que o uso concomitante de metotrexato e outros imunomoduladores em pacientes com artrite reumatóide, artrite psoriática e doença de Crohn reduz a formação de anticorpos contra infliximabe e aumenta as concentrações plasmáticas de infliximabe. No entanto, os resultados são incertos devido às limitações dos métodos usados para a dosagem do infliximabe e dos anticorpos anti-infliximabe no soro.
Os corticosteróides não parecem alterar a farmacocinética do infliximab de uma forma clinicamente relevante.
A combinação de Remicade com outras terapias biológicas utilizadas para tratar as mesmas condições que Remicade, incluindo anakinra e abatacept, não é recomendada (ver secção 4.4).
Recomenda-se que as vacinas vivas não sejam administradas ao mesmo tempo que o Remicade. Também é recomendado que vacinas vivas não sejam administradas a bebês após a exposição no utero ao infliximab durante pelo menos 6 meses após o nascimento (ver secção 4.4).
Agentes terapêuticos infecciosos não devem ser administrados concomitantemente com Remicade (ver secção 4.4).
04.6 Gravidez e lactação
Mulheres em idade fértil
As mulheres com potencial para engravidar devem usar métodos contracetivos adequados durante o tratamento com Remicade e continuar a sua utilização pelo menos 6 meses após a última dose.
Gravidez
Um número moderado de dados coletados prospectivamente em pacientes grávidas (aproximadamente 450) tratadas com infliximabe com resultados conhecidos, incluindo um número limitado (aproximadamente 230) de gravidezes tratadas durante o primeiro trimestre, não mostraram efeitos inesperados no resultado. Devido à inibição do TNFα, infliximabe administrado durante a gravidez pode alterar as respostas imunológicas normais do recém-nascido. Nem toxicidade materna, embriotoxicidade, nem teratogenicidade foram encontradas em um estudo de toxicidade de desenvolvimento em camundongos usando um anticorpo semelhante que inibe seletivamente a funcionalidade do TNFα (ver seção 5.3).
A experiência clínica disponível é muito limitada para excluir riscos e, portanto, a administração de infliximabe não é recomendada durante a gravidez.
O infliximabe passa pela placenta e foi detectado no soro de bebês por até 6 meses após o nascimento. Após a exposição no utero ao infliximabe, os bebês podem ter um risco maior de infecção, incluindo uma "infecção disseminada grave que pode ter um resultado fatal. A administração de vacinas vivas (por exemplo, vacina BCG) a bebês expostos no utero infliximab não é recomendado durante pelo menos 6 meses após o nascimento (ver secções 4.4 e 4.5). Também foram notificados casos de agranulocitose (ver secção 4.8).
Hora da alimentação
Não se sabe se o infliximab é excretado no leite materno ou absorvido sistemicamente após a ingestão.Uma vez que as imunoglobulinas humanas são excretadas no leite materno, as mulheres não devem amamentar durante pelo menos 6 meses após o tratamento com Remicade.
Fertilidade
Não existem dados pré-clínicos suficientes para tirar conclusões sobre os efeitos do infliximab na fertilidade e na função reprodutiva global (ver secção 5.3).
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
Remicade tem efeitos menores sobre a capacidade de conduzir ou utilizar máquinas. Podem ocorrer tonturas após a administração de Remicade (ver secção 4.8).
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
A infecção do trato respiratório superior foi a reação adversa mais comum (RAM) relatada em ensaios clínicos, ocorrendo em 25,3% dos pacientes tratados com infliximabe em comparação com 16,5% dos pacientes controle. (CHF), infecções graves (incluindo sepse, infecções oportunistas e TB), doença do soro (reações de hipersensibilidade retardada), reações hematológicas, lúpus eritematoso sistêmico / síndrome semelhante ao lúpus, doença desmielinizante, eventos hepatobiliares, linfoma, HSTCL, leucemia, célula de Merkel carcinoma, melanoma, malignidade pediátrica, sarcoidose / reação do tipo sarcoide, abcesso intestinal ou perianal (na doença de Crohn) e reações graves à perfusão (ver secção 4.4).
Tabela de reações adversas
A Tabela 1 lista as RAMs notificadas em ensaios clínicos, bem como as reações adversas, algumas com resultado fatal, notificadas após a comercialização. Dentro da classe de sistemas de órgãos, as reações adversas são listadas por frequência usando as seguintes categorias: muito frequentes (≥ 1/10); frequentes (≥ 1/100 a
tabela 1
Efeitos indesejáveis em ensaios clínicos e após comercialização
* incluindo tuberculose bovina (infecção disseminada por BCG), ver seção 4.4
Reações relacionadas à infusão
Em estudos clínicos, uma reação relacionada à infusão foi definida como qualquer evento adverso que ocorre durante uma infusão ou dentro de 1 hora após a infusão. Em estudos clínicos de fase III, 18% dos pacientes tratados com infliximabe em comparação com 5% dos pacientes tratados com placebo tiveram uma reação relacionada à infusão. No geral, uma proporção maior de pacientes que receberam monoterapia com infliximabe experimentou uma reação relacionada à infusão do que pacientes que receberam infliximabe concomitante com imunomoduladores. Aproximadamente 3% dos pacientes interromperam o tratamento devido a reações relacionadas à infusão e todos os pacientes se recuperaram com ou sem terapia médica.
Dos pacientes tratados com infliximabe que tiveram uma reação à infusão durante o período de indução até a semana 6, 27% experimentaram uma reação à infusão durante o período de manutenção entre a semana 7 e a semana 54. Dos pacientes que não tiveram uma reação à infusão durante o período de indução, 9% experimentaram uma reação à infusão durante o período de manutenção.
Em um estudo clínico em pacientes com artrite reumatóide (ASPIRE), as infusões foram administradas durante 2 horas nas primeiras 3 infusões. A duração das infusões subsequentes pode ser encurtada para não menos de 40 minutos em pacientes que não apresentaram quaisquer reações. a infusão. Neste estudo, sessenta e seis por cento dos pacientes (686 em 1.040) receberam pelo menos uma infusão abreviada com duração de 90 minutos ou menos e 44% dos pacientes (454 em 1.040) receberam pelo menos uma infusão abreviada com duração de 60 minutos ou menos. Em pacientes tratados com infliximabe que receberam pelo menos uma infusão abreviada, reações relacionadas à infusão ocorreram em 15% dos pacientes e reações graves à infusão ocorreram em 0,4% dos pacientes.
Em um estudo clínico em pacientes com doença de Crohn (SONIC), reações relacionadas à infusão foram observadas em 16,6% (27/163) dos pacientes recebendo infliximabe em monoterapia, em 5% (9/179) dos pacientes recebendo infliximabe em monoterapia. Pacientes recebendo infliximabe em combinação com AZA e em 5,6% (9/161) dos pacientes recebendo monoterapia com AZA. Uma reação grave à infusão (
No período pós-comercialização, foram associados à administração de Remicade casos de reações anafilactóides, incluindo edema laríngeo / faríngeo, broncoespasmo grave e convulsões.
Além disso, também houve notificações raras de perda transitória da visão e isquemia do miocárdio / enfarte do miocárdio ocorridos durante ou nas duas horas após a perfusão de Remicade (ver secção 4.4).
Reações à infusão após a re-administração de Remicade
Um estudo clínico foi desenhado em pacientes com psoríase moderada a grave para avaliar a eficácia e segurança da terapia de manutenção de longo prazo, em comparação com o retratamento com um regime de indução de Remicade (máximo de quatro infusões em 0, 2, 6 e 14 semanas) após a doença Os pacientes não receberam terapia imunossupressora concomitante. No braço do retratamento, 4% (8/219) dos pacientes apresentaram reações graves à infusão de edema facial e hipotensão. Em todos os casos, o tratamento com Remicade foi interrompido e / ou adotado outro tratamento com resolução completa dos sinais e sintomas.
Hipersensibilidade retardada
Em estudos clínicos, as reações de hipersensibilidade retardada foram pouco frequentes e ocorreram após intervalos de tempo sem Remicade inferiores a 1 ano. Em estudos de psoríase, reações de hipersensibilidade tardia ocorreram no início do tratamento. Os sinais e sintomas incluíram mialgia e / ou artralgia com febre e / ou erupção cutânea, com alguns pacientes apresentando prurido, edema facial, de mãos ou lábios, disfagia, urticária, dor de garganta e cefaleia.
Não existem dados suficientes sobre a incidência de reações de hipersensibilidade retardada após intervalos de tempo sem Remicade de mais de 1 ano, mas dados limitados de ensaios clínicos sugerem um aumento do risco de hipersensibilidade retardada ao aumento. A duração dos intervalos de tempo sem administração de Remicade (ver seção 4.4).
Num estudo clínico de 1 ano com perfusões repetidas em doentes com doença de Crohn (estudo ACCENT I), a incidência de reacções resultantes do desenvolvimento de reacções semelhantes à doença do soro foi de 2,4%.
Imunogenicidade
Os doentes que desenvolveram anticorpos ao infliximab tinham maior probabilidade de apresentar reações relacionadas com a perfusão (aproximadamente 2 a 3 vezes mais) .O uso concomitante de agentes imunossupressores pareceu reduzir a frequência das reações relacionadas com a perfusão.
Em ensaios clínicos em que doses únicas e múltiplas de infliximabe foram administradas variando de 1 a 20 mg / kg, anticorpos para infliximabe foram encontrados em 14% dos pacientes recebendo qualquer terapia imunossupressora e em 24% dos pacientes sem terapia imunossupressora. 8% dos pacientes com artrite reumatoide tratados repetidamente com a dosagem recomendada e metotrexato desenvolveram anticorpos contra o infliximabe. Em pacientes com artrite psoriática tratados com 5 mg / kg com ou sem metotrexato, os anticorpos se desenvolveram em 15% do total dos pacientes (em 4% dos pacientes que receberam metotrexato e 26% dos pacientes que não receberam metotrexato no início do estudo). Em pacientes com doença de Crohn recebendo tratamento de manutenção, em média 3,3% dos pacientes que receberam imunossupressores e 13,3% dos pacientes que não receberam imunossupressores desenvolveram anticorpos contra o infliximabe. A incidência de anticorpos foi 2-3 vezes maior para pacientes tratados episodicamente. Devido a limitações metodológicas, um teste negativo não excluiu a presença de anticorpos contra o infliximabe. Alguns pacientes que desenvolveram altos títulos de anticorpos contra o infliximabe tiveram eficácia reduzida. Em doentes com psoríase tratados com regime de manutenção com infliximab, na ausência de tratamento concomitante com imunomoduladores, aproximadamente 28% desenvolveram anticorpos contra o infliximab (ver secção 4.4: “Reacções à perfusão e hipersensibilidade”).
Infecções
Tuberculose, infecções bacterianas, incluindo sepsia e pneumonia, infecções invasivas fúngicas, virais e outras infecções oportunistas, foram observadas em doentes a receber Remicade. Alguns deles foram fatais. As infecções oportunistas notificadas com mais frequência com uma taxa de mortalidade> 5% incluem pneumocistose, candidíase, listeriose e aspergilose (ver secção 4.4).
Em ensaios clínicos, 36% dos pacientes tratados com infliximabe foram tratados para infecções, em comparação com 25% dos pacientes tratados com placebo.
Em ensaios clínicos com artrite reumatóide, a incidência de infecções graves, incluindo pneumonia, foi superior em doentes tratados com infliximab e metotrexato do que naqueles tratados apenas com metotrexato, especialmente em doses de 6 mg / kg ou superiores (ver secção 4.4).
Entre as notificações espontâneas notificadas no período pós-comercialização, as infecções são o efeito adverso grave mais comum. Alguns dos casos tiveram um resultado fatal. Quase 50% das mortes notificadas foram associadas a infecções. Foram notificados casos de tuberculose., por vezes fatal, incluindo casos de tuberculose miliar e tuberculose de localização extrapulmonar (ver secção 4.4).
Neoplasias malignas e doenças linfoproliferativas
Em ensaios clínicos realizados com infliximab nos quais 5.780 pacientes, representando 5.494 pacientes-ano, foram tratados, foram detectados 5 casos de linfomas e 26 casos de neoplasias não linfáticas, em comparação com nenhum caso de linfoma e 1 caso de neoplasias não linfáticas. observado em 1.600 pacientes tratados com placebo, representando 941 pacientes-ano.
Em estudos clínicos de segurança de longo prazo de até 5 anos com infliximabe, representando 6.234 pacientes-ano (3.210 pacientes), foram relatados 5 casos de linfoma e 38 casos de neoplasias não linfáticas.
Também foram notificados casos de doenças malignas, incluindo linfoma, durante o período pós-comercialização (ver secção 4.4).
Num ensaio clínico exploratório envolvendo doentes com DPOC moderada a grave que eram fumadores ou ex-fumadores, 157 doentes adultos foram tratados com Remicade em doses semelhantes às utilizadas na artrite reumatóide e na doença de Crohn. Nove desses pacientes desenvolveram doenças malignas, incluindo 1 linfoma. A duração mediana de um acompanhamento foi de 0,8 anos (incidência 5,7% [IC 95% 2,65% - 10,6%]. Um caso de malignidade foi relatado entre os 77 pacientes no controle (duração mediana de acompanhamento 0,8 anos; incidência 1,3% [IC 95% 0,03% - 7,0%]). A maioria dessas doenças malignas envolveu pulmão, cabeça ou pescoço.
Um estudo de coorte retrospectivo de base populacional encontrou um aumento na incidência de câncer cervical em mulheres tratadas com infliximabe com artrite reumatóide em comparação com pacientes não tratados biologicamente ou na população em geral, incluindo aquelas com mais de 60 anos de idade (ver seção 4.4).
Além disso, foram notificados casos raros de linfoma hepatoesplénico das células T após a comercialização em doentes tratados com Remicade, a grande maioria dos casos ocorreu em doentes com doença de Crohn e colite ulcerosa, a maioria dos doentes eram adolescentes ou jovens adultos do sexo masculino (ver secção 4.4 )
Insuficiência cardíaca
Em um estudo de fase II com o objetivo de avaliar Remicade na Insuficiência Cardíaca Congestiva (ICC), foi encontrada maior incidência de mortalidade por agravamento da insuficiência cardíaca em pacientes tratados com Remicade, principalmente naqueles tratados. Com a dose mais alta de 10 mg / kg (ou seja, o dobro da dose máxima aprovada). Neste estudo, 150 pacientes com ICC classe III e IV da NYHA (fração de ejeção do ventrículo esquerdo ≤ 35%) foram tratados com 3 infusões de Remicade 5 mg / kg, 10 mg / kg ou placebo durante um período de 6 semanas. Na 38ª semana, 9 dos 101 pacientes tratados com Remicade (2 a 5 mg / kg e 7 a 10 mg / kg) morreram enquanto houve uma morte entre os 49 pacientes tratados com placebo.
Casos de agravamento da insuficiência cardíaca, com e sem desencadeadores identificáveis, foram relatados durante o período pós-comercialização em pacientes tratados com Remicade. Novo início de insuficiência cardíaca, incluindo insuficiência cardíaca, também foi relatado durante o período pós-comercialização. nenhuma doença cardiovascular preexistente conhecida Alguns desses pacientes tinham menos de 50 anos de idade.
Eventos hepatobiliares
Em estudos clínicos, foram observadas elevações ligeiras ou moderadas nas ALT e AST em doentes a receber Remicade sem progredir para lesão hepática grave. Foram observadas elevações de ALT ≥ 5 x acima dos limites normais (ULN) (consulte a Tabela 2). Elevações da aminotransferase (mais comuns em ALT do que em AST) foram observadas em uma proporção maior de pacientes que receberam Remicade do que nos grupos de controle, tanto quando Remicade foi administrado sozinho quanto quando administrado em combinação com outras drogas imunossupressoras. A maioria das anormalidades da aminotransferase foram transitórias; no entanto, aumentos prolongados ocorreram em um pequeno número de pacientes. Em geral, os doentes que desenvolveram elevações nas ALT e AST eram assintomáticos e as anomalias diminuíram ou foram resolvidas continuando ou descontinuando o tratamento com Remicade ou alterando a terapia concomitante. Foram notificados casos muito raros de icterícia e hepatite, alguns com características de hepatite autoimune, em doentes a receber Remicade durante o período de vigilância pós-comercialização (ver secção 4.4).
mesa 2
Número de pacientes com aumento da atividade de ALT em ensaios clínicos
1 Os pacientes do grupo placebo receberam metotrexato, enquanto os pacientes do grupo infliximabe receberam infliximabe e metotrexato.
2 Os pacientes no grupo de placebo nos 2 estudos de Fase III da doença de Crohn, ACCENT I e ACCENT II, receberam uma dose inicial de 5 mg / kg de infliximabe no início do estudo e placebo na fase de manutenção. Foram randomizados para o grupo de manutenção com placebo. e posteriormente mudados para infliximabe, eles foram incluídos no grupo do infliximabe na análise de ALT. No estudo de Fase IIIb na doença de Crohn, SONIC, os pacientes no braço do placebo receberam AZA 2,5 mg / kg / dia como controle ativo, além de infusões de infliximabe com placebo.
3 Número de pacientes avaliados para ALT.
4 O seguimento médio é baseado nos pacientes tratados.
Anticorpos antinucleares (ANA) / anticorpos de DNA de fita dupla (dsDNA)
Cerca de metade dos pacientes tratados com infliximabe em ensaios clínicos que eram ANA negativos no início do estudo tornaram-se ANA positivos durante o estudo, em comparação com cerca de um quinto dos pacientes tratados com placebo. Anticorpos anti-dsDNA foram detectados recentemente em aproximadamente 17% dos pacientes tratados com infliximabe em comparação com 0% dos pacientes tratados com placebo. Na avaliação mais recente, 57% dos doentes tratados com infliximab permaneceram positivos para anticorpos anti-dsDNA.No entanto, as notificações de lúpus e síndromes lúpicas semelhantes permanecem pouco frequentes (ver secção 4.4).
População pediátrica
Pacientes com artrite reumatóide juvenil
Remicade foi estudado em um estudo clínico envolvendo 120 pacientes (faixa etária: 4-17 anos) com artrite reumatóide juvenil ativa, independentemente do uso de metotrexato. Os pacientes foram tratados com infliximabe 3 ou 6 mg / kg como um regime de indução de 3 doses (semana 0 , 2, 6 ou semanas 14,16, 20 respectivamente) seguido por terapia de manutenção a cada 8 semanas, em combinação com metotrexato.
Reações de infusão
As reações à infusão ocorreram em 35% dos pacientes com artrite reumatóide juvenil que receberam 3 mg / kg em comparação com 17,5% dos pacientes que receberam 6 mg / kg. No grupo de Remicade 3 mg / kg, 4 dos 60 pacientes tiveram uma reação grave à infusão e 3 pacientes relataram uma possível reação anafilática (2 das quais foram incluídas em reações graves à perfusão). No grupo de 6 mg / kg, 2 de 57 doentes experimentaram uma reação grave à perfusão. "perfusão, uma das quais teve uma possível reação anafilática (ver secção 4.4) .
Imunogenicidade
38% dos pacientes que receberam 3 mg / kg desenvolveram anticorpos contra o infliximabe em comparação com 12% dos pacientes que receberam 6 mg / kg. Os títulos de anticorpos foram significativamente maiores no grupo que recebeu 3 mg / kg do que no grupo que recebeu 6 mg / kg.
Infecções
As infecções ocorreram em 68% (41/60) das crianças que receberam 3 mg / kg por 52 semanas, em 65% (37/57) das crianças que receberam 6 mg / kg de infliximabe por 38 semanas e em 47% (28 / 60) de crianças que receberam placebo durante 14 semanas (ver secção 4.4).
Pacientes pediátricos com doença de Crohn
Os seguintes efeitos indesejáveis foram notificados mais frequentemente em doentes pediátricos com doença de Crohn incluídos no estudo REACH (ver secção 5.1) do que em doentes adultos com doença de Crohn: anemia (10,7%), sangue nas fezes (9,7%), leucopenia (8,7%), rubor com vermelhidão da pele (8,7%), infecções virais (7,8%), neutropenia (6,8%), fraturas ósseas (6,8%), infecções bacterianas (5,8%) e reações alérgicas envolvendo o trato respiratório (5,8%). Outras considerações especiais são descritas abaixo.
Reações relacionadas à infusão
17,5% dos pacientes randomizados no estudo REACH apresentaram 1 ou mais reações à infusão. Não foram relatados casos graves de reações à infusão e 2 participantes no estudo REACH desenvolveram reações anafiláticas não graves.
Imunogenicidade
Anticorpos para infliximabe foram detectados em 3 (2,9%) dos pacientes pediátricos.
Infecções
No estudo REACH, infecções foram relatadas em 56,3% dos indivíduos randomizados tratados com infliximabe. As infecções foram relatadas com mais frequência em indivíduos que receberam infusões a cada 8 semanas do que naqueles tratados a cada 12 semanas (73,6% e 38,0%, respectivamente), enquanto infecções graves foram relatadas em 3 indivíduos no grupo de tratamento de manutenção a cada 8 semanas e em 4 indivíduos em o grupo tratado a cada 12 semanas. As infecções mais comumente relatadas foram infecção do trato respiratório superior e faringite. O abcesso foi a infecção grave mais comum. Foram relatados 3 casos de pneumonia (1 grave) e 2 casos de herpes zoster (ambos não graves).
Pacientes pediátricos com colite ulcerosa
No geral, as reações adversas notificadas no estudo em doentes pediátricos com colite ulcerosa (C0168T72) foram geralmente consistentes com as notificadas em estudos em doentes adultos com colite ulcerosa (ACT 1 e ACT 2). No estudo C0168T72, as reações adversas mais comuns foram infecção do trato respiratório superior, faringite, dor abdominal, febre e dor de cabeça. O evento adverso mais comum foi o agravamento da colite ulcerosa, cuja incidência foi maior em pacientes tratados a cada 12 semanas do que naqueles tratados a cada 8 semanas.
Reações relacionadas à infusão
No geral, 8 (13,3%) dos 60 pacientes tratados relataram uma ou mais reações à infusão, com 4 dos 22 pacientes (18,2%) no grupo de manutenção tratados a cada 8 semanas e 3 dos 23 pacientes (13, 0%) ) no grupo de manutenção tratado a cada 12 semanas Não foram relatadas reações graves à perfusão. Todas as reações à infusão foram de intensidade leve ou moderada.
Imunogenicidade
Anticorpos para infliximabe foram detectados em 4 (7,7%) dos pacientes até a semana 54.
Infecções
Infecções foram relatadas em 31 (51,7%) dos 60 pacientes tratados no estudo C0168T72 e 22 (36,7%) necessitaram de tratamento antimicrobiano oral ou parenteral. A proporção de doentes com infecções no estudo C0168T72 foi semelhante à do estudo pediátrico da doença de Crohn (REACH), mas superior à proporção dos estudos de colite ulcerosa em adultos (ACT 1 e ACT 2). A incidência geral de infecções no estudo C0168T72 foi de 13/22 (59%) no grupo de manutenção tratado a cada 8 semanas e 14/23 (60,9%) no grupo de manutenção tratado a cada 12 semanas. Trato respiratório superior (7/60 [12 %]) e faringite (5/60 [8%]) foram as infecções do sistema respiratório mais frequentemente notificadas. Infecções graves foram relatadas em 12% (7/60) de todos os pacientes tratados.
Neste estudo, havia mais pacientes na faixa etária de 12 a 17 anos do que na faixa de 6 a 11 anos (45/60 [75,0%]) vs. 15/60 [25,0%]). Embora o número de pacientes em cada subgrupo fosse muito pequeno para tirar quaisquer conclusões firmes sobre o "efeito da idade nos eventos relacionados à segurança", houve uma proporção maior de pacientes com eventos adversos graves e descontinuação do tratamento causados por eventos. Adverso nos mais jovens. faixa etária do que na faixa etária mais velha. Embora a proporção de pacientes com infecções também tenha sido maior na faixa etária mais jovem, para infecções graves, as proporções nos dois grupos foram semelhantes. A proporção geral de eventos adversos e reações à infusão foi semelhante entre as faixas etárias de 6 a 11 anos e de 12 a 17 anos.
Experiência pós-marketing
A notificação espontânea pós-comercialização de acontecimentos adversos graves em doentes pediátricos incluiu doenças malignas incluindo linfoma hepatoesplénico das células T, anomalias transitórias das enzimas hepáticas, síndromes semelhantes ao lúpus e autoanticorpos positivos (ver secções 4.4 e 4.8).
Informações adicionais sobre populações especiais
Pacientes idosos (≥ 65 anos)
Em ensaios clínicos de artrite reumatóide, a incidência de infecções graves foi maior em pacientes tratados com infliximabe mais metotrexato com 65 anos ou mais (11,3%) do que em pacientes com menos de 65 anos (4, 6%). Em doentes tratados apenas com metotrexato, a incidência de infecções graves foi de 5,2% em doentes com 65 ou mais anos, em comparação com 2,7% em doentes com menos de 65 anos (ver secção 4.4).
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas que ocorram após a autorização do medicamento é importante, uma vez que permite a monitorização contínua da relação benefício / risco do medicamento.Os profissionais de saúde são convidados a notificar quaisquer suspeitas de reações adversas à Agência Italiana de Medicamentos. , site: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdose
Nenhum caso de sobredosagem foi relatado. Doses únicas até um máximo de 20 mg / kg foram administradas sem efeitos tóxicos.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: inibidores do fator de necrose tumoral alfa (TNF-alfa).
Código ATC: L04AB02.
Mecanismo de ação
O infliximabe é um anticorpo monoclonal quimérico, humano-murino, que se liga com alta afinidade às formas solúvel e transmembrana do TNFα, mas não à linfotoxina α (TNFβ).
Efeitos farmacodinâmicos
Infliximabe inibe em vitro Atividade do TNFα em uma ampla gama de dosagens biológicas. O infliximabe preveniu a doença em camundongos transgênicos que desenvolvem poliartrite como consequência da expressão humana essencial do TNFα e, quando administrado após o início da doença, permitiu a regressão das erosões articulares. Na Vivo, o infliximabe forma rapidamente complexos estáveis com o TNFα humano, um processo que leva à perda da atividade biológica do TNFα.
Altas concentrações de TNFα foram detectadas nas articulações de pacientes com artrite reumatoide e relacionadas à alta atividade da doença. O tratamento com infliximabe resultou na redução da infiltração de células inflamatórias nas áreas inflamadas das articulações e na redução da expressão na artrite reumatóide, moléculas que medeiam a adesão celular, quimiotaxia e degradação do tecido. Após o tratamento com infliximabe, os pacientes apresentaram níveis séricos reduzidos de interleucina 6 (IL-6) e proteína C reativa (PCR) e níveis elevados de hemoglobina em pacientes com AR com níveis de hemoglobina reduzidos em comparação com os valores pré-tratamento. Além disso, os linfócitos do sangue periférico não mostraram uma diminuição significativa no número e na resposta proliferativa ao teste em vitro da estimulação mitogênica em comparação com as células de pacientes não tratados. Em pacientes com psoríase, o tratamento com infliximabe resultou na diminuição da inflamação epidérmica e normalização da diferenciação dos queratinócitos em placas psoriáticas. Na artrite psoriática, o tratamento de curto prazo com Remicade reduziu o número de células T e vasos sanguíneos na sinóvia e na pele psoriática.
Uma avaliação histológica de biópsias de cólon realizadas antes e 4 semanas após a administração de infliximabe revelou uma redução substancial no TNFα detectável. O tratamento com infliximabe de pacientes com doença de Crohn também foi associado a uma substancial
diminuição da concentração sérica de PCR, um marcador inflamatório comumente elevado. A contagem total de leucócitos periféricos foi minimamente afetada em pacientes tratados com infliximabe, embora as alterações nos linfócitos, monócitos e neutrófilos refletissem alterações dos valores normais. As células mononucleares do sangue periférico (PBMCs) de pacientes tratados com infliximabe mostraram uma capacidade de resposta proliferativa não afetada aos estímulos, em comparação com pacientes não tratados; e, além disso, após o tratamento com infliximabe, nenhuma mudança substancial foi observada na produção de citocinas pelas células PBMC estimuladas. A análise das células mononucleares da lâmina própria obtidas após biópsia da mucosa intestinal mostrou que o tratamento com infliximabe resultou na redução do número de células capazes de expressar TNFα e interferon γ. Outros estudos histológicos forneceram evidências de que o tratamento com infliximabe reduz a infiltração de células inflamatórias nas áreas envolvidas do intestino e a presença de marcadorEstudos endoscópicos da mucosa intestinal mostraram cicatrização da mucosa em pacientes tratados com infliximabe.
Eficácia clínica e segurança
Artrite reumatóide em adultos
A eficácia do infliximabe foi avaliada em dois ensaios clínicos piloto multicêntricos, randomizados e duplo-cegos: ATTRACT e ASPIRE. Em ambos os estudos, o uso concomitante de doses estáveis de ácido fólico, corticosteroides orais (≤ 10 mg / dia) e / ou não antiinflamatórios esteróides (AINEs).
Os desfechos primários foram redução de sinais e sintomas conforme definido pelos critérios do American College of Rheumatology (ACR20 para ATTRACT, indicador ACR-N para ASPIRE), prevenção de danos estruturais nas articulações e melhora na função física.Uma redução nos sinais e sintomas foi definida como uma melhora de pelo menos 20% (ACR20) no número de articulações doloridas e inchadas e em 3 dos 5 critérios a seguir: Avaliação Global do Médico, Avaliação Global do Paciente, Avaliação Funcional / deficiência, dor escala visual analógica, taxa de hemossedimentação ou proteína C reativa. O ACR-N usa os mesmos critérios do ACR20, calculado considerando o menor percentual de melhora na contagem de articulações inchadas, articulações doloridas e a mediana dos 5 componentes restantes da resposta ACR. Articulação) em ambas as mãos e pés, foi medido por avaliar a mudança da linha de base na pontuação de Sharp total modificada de van der Heijde (0-440) .O Health Assessment Questionnaire (HAQ; escala de 0 a 3) foi usado para avaliar a mudança média na função física ao longo do tempo desde a linha de base.
O estudo ATTRACT avaliou as respostas nas semanas 30, 54 e 102 em um estudo controlado com placebo em 428 pacientes com artrite reumatóide ativa, apesar do tratamento com metotrexato. Aproximadamente 50% dos pacientes estavam em classe funcional III. Os pacientes foram tratados com placebo, 3 mg / kg ou 10 mg / kg de infliximabe nas semanas 0, 2 e 6 e a cada 4 ou 8 semanas daí em diante. Todos os pacientes tomaram uma dose estável de metotrexato (mediana de 15 mg / semana) por 6 meses antes da inscrição e permaneceram em doses estáveis ao longo do estudo.
Os resultados na semana 54 (ACR20, pontuação total de Sharp modificada por van der Heijde e HAQ) são mostrados na Tabela 3. Uma maior incidência de resposta clínica (ACR50 e ACR70) foi observada em todos os grupos tratados com infliximabe nas semanas 30 e 54 em comparação com metotrexato sozinho.
Uma redução na taxa de progressão dos danos estruturais da articulação (erosão e redução da lacuna articular) foi observada em todos os grupos tratados com infliximabe em 54 semanas (Tabela 3).
Os efeitos observados na semana 54 foram mantidos até a semana 102 do tratamento. Devido ao número de interrupções do tratamento, a extensão da diferença no efeito entre os grupos de monoterapia com infliximabe e metotrexato não pôde ser definida.
Tabela 3
Efeitos no ACR20, dano estrutural da articulação e função física na semana 54, ATRAIR
controlado = Todos os pacientes tinham AR ativa, apesar do tratamento com doses estáveis de metotrexato por 6 meses antes da inscrição e tiveram que permanecer em doses estáveis durante o estudo. Uso concomitante de doses estáveis de corticosteroides orais (≤ 10 mg / dia) e / ou a medicamento antiinflamatório não esteroidal (AINE) foi permitido; um suplemento de folato foi dado.
b Todas as doses de infliximabe foram administradas concomitantemente com metotrexato e folato e, em alguns casos, com corticosteroides e / ou antiinflamatórios não esteroidais (AINEs)
c p
d valores mais altos indicam maior dano articular.
e HAQ = Questionário de Avaliação de Saúde; valores mais altos indicam uma "deficiência menor.
O estudo ASPIRE avaliou as respostas na semana 54 em 1.004 pacientes previamente virgens de metotrexato com artrite reumatóide ativa (contagem média de articulações inchadas e doloridas: 19 e 31, respectivamente) de início recente (duração da doença ≤ 3 anos, mediana de 0,6 anos). Todos os pacientes receberam metotrexato (otimizado para 20 mg / semana na semana 8) em combinação com placebo ou infliximabe 3 mg / kg ou 6 mg / kg nas semanas 0, 2 e 6 e a cada 8 semanas daí em diante. Os resultados na semana 54 são mostrados na Tabela 4.
Após 54 semanas de tratamento, ambas as doses de infliximabe + metotrexato deram uma melhora estatisticamente significativa nos sinais e sintomas maiores do que o metotrexato sozinho, conforme medido pela proporção de pacientes que alcançaram respostas ACR 20, 50 e 70.
No ASPIRE, mais de 90% dos pacientes tinham pelo menos duas radiografias avaliáveis. A redução na taxa de progressão dos danos estruturais foi observada nas semanas 30 e 54 nos grupos infliximabe + metotrexato em comparação com metotrexato sozinho.
Tabela 4
Efeitos no ACRn, danos estruturais nas articulações e função física na semana 54, ASPIRE
no p
b valores mais altos indicam maior dano articular.
c questionário de avaliação de saúde; valores mais altos indicam uma "deficiência menor.
d p = 0,030 e
Os dados para apoiar a titulação da dose na artrite reumatóide vêm dos estudos
ATRAIR, ASPIRE e INICIAR. O START foi um estudo de segurança randomizado, multicêntrico, duplo-cego, de três braços e de grupos paralelos. Em um dos braços do estudo (grupo 2, n = 329), os pacientes com resposta inadequada foram autorizados a titulação da dose em aumentos de 1,5 mg / kg de 3 a 9 mg / kg. A maioria (67%) desses pacientes não precisou de titulação da dose. Dos pacientes que necessitaram, 80% obtiveram resposta clínica e a maioria (64%) destes necessitou apenas de um aumento de 1,5 mg / kg.
Doença de Crohn em adultos
Tratamento de indução na doença de Crohn ativa, moderada a grave A eficácia de uma dose única de infliximabe foi avaliada em 108 pacientes com doença de Crohn ativa variando em gravidade de moderada a grave (índice de atividade da doença de Crohn (CDAI) ≥ 220 ≤ 400) em um estudo randomizado, duplo-cego, dose controlada por placebo resposta. Dos 108 pacientes, 27 foram tratados com a dosagem recomendada de infliximabe (5 mg / kg). Todos os pacientes tiveram resposta inadequada a terapias convencionais anteriores O uso concomitante de doses inalteradas de terapias convencionais foi permitido e 92% dos pacientes então continuaram a receber tais terapias.
O endpoint primário foi o cálculo do número de pacientes que experimentaram uma resposta clínica, definida como uma diminuição no CDAI de ≥ 70 pontos da linha de base, na semana 4 e sem aumento no uso de medicamentos ou cirurgia para a doença. Os pacientes que responderam na semana 4 foram acompanhados até a semana 12. Os desfechos secundários incluíram o número de pacientes em remissão clínica na semana 4 (CDAI
Na semana 4, após a administração de dose única, 22/27 (81%) pacientes tratados com infliximabe na dose de 5 mg / kg tiveram uma resposta clínica em comparação com 4/25 (16%) pacientes tratados com placebo (p
Tratamento de manutenção na doença de Crohn ativa, moderada a grave em adultos
A eficácia de infusões repetidas com infliximabe foi avaliada em um estudo clínico de 1 ano (ACCENT I). Um total de 573 pacientes com doença de Crohn ativa moderada a grave (CDAI ≥ 220 ≤ 400) receberam uma única infusão de 5 mg / kg em semana 0. 178 dos 580 pacientes inscritos (30,7%) tinham doença grave (pontuação CDAI> 300 e terapia concomitante com corticosteroides e / ou imunossupressores) correspondendo à população definida nas indicações (ver seção 4.1) Na semana 2, todos os pacientes foram avaliados quanto à resposta clínica e randomizados em um dos 3 grupos de tratamento; um grupo de manutenção com placebo, um grupo de manutenção com 5 mg / kg e uma manutenção com 10 mg / kg. Em seguida, todos os 3 grupos receberam infusões repetidas nas semanas 2, 6 e a cada 8 semanas depois.
Dos 573 pacientes randomizados, 335 (58%) obtiveram resposta clínica na semana 2. Esses pacientes foram classificados como respondedores da semana 2 e incluídos na análise primária (ver Tabela 5). Semana 2, 32% (26/81) no o grupo de manutenção com placebo e 42% (68/163) no grupo de manutenção com infliximabe obtiveram resposta clínica na semana 6. Depois disso, não houve diferença entre os grupos no número de pacientes que subsequentemente responderam à terapia.
Os desfechos co-primários foram a porcentagem de pacientes em remissão clínica (CDAI
Tabela 5
Efeitos na velocidade de resposta e remissão, dados do ACCENT I (pacientes que alcançaram a resposta na semana 2)
a Redução do CDAI ≥ 25% e ≥ 70 pontos.
b CDAI
No início da semana 14, os pacientes que responderam ao tratamento, mas posteriormente perderam o benefício clínico, foram trocados para uma dose de infliximabe 5 mg / kg maior do que a dose para a qual foram originalmente randomizados. L "Oitenta e nove por cento (50 / 56) dos pacientes que perderam a resposta clínica à terapia de manutenção com infliximabe 5 mg / kg após a semana 14 responderam ao tratamento com infliximabe 10 mg / kg.
Nas semanas 30 e 54, melhorias nas avaliações da qualidade de vida, uma redução nas hospitalizações relacionadas à doença e o uso de corticosteroides foram observados nos grupos de manutenção com infliximabe em comparação com o grupo de manutenção com placebo.
O infliximabe, com ou sem AZA, foi avaliado em um estudo randomizado, duplo-cego, comparador ativo (SONIC) de 508 pacientes adultos com doença de Crohn moderada a grave (CDAI ≥ 220 ≤ 450) que nunca haviam sido tratados antes. Com produtos biológicos e imunossupressores e com uma duração mediana da doença de 2,3 anos. No início do estudo, 27,4% dos pacientes estavam em corticosteroides sistêmicos, 14,2% dos pacientes em budesonida e 54,3% dos pacientes em compostos 5-ASA. Os pacientes foram randomizados para receber monoterapia com AZA, monoterapia com infliximabe ou terapia combinada com infliximabe e AZA. O infliximabe foi administrado na dose de 5 mg / kg nas semanas 0, 2, 6 e a cada 8 semanas daí em diante. AZA foi administrado na dose diária de 2,5 mg / kg.
O endpoint primário do estudo foi remissão clínica sem corticosteroides na semana 26, definida como pacientes em remissão clínica (prednisona CDAI ou equivalente) ou budesonida em dose> 6 mg / dia. Para resultados, consulte a Tabela 6. Proporções de pacientes com cicatrização da mucosa na semana 26 foram significativamente maiores nos grupos de combinação de infliximabe e AZA (43,9%, p
Tabela 6
Porcentagem de pacientes que alcançaram remissão clínica sem corticosteroides na Semana 26, SONIC
* Os valores P representam cada grupo de tratamento com infliximabe versus monoterapia com AZA
Padrões semelhantes na obtenção de remissão clínica sem corticosteroides foram observados na Semana 50. Além disso, uma melhora na qualidade de vida foi observada com infliximabe, conforme relatado pelo questionário IBDQ.
Tratamento de indução na doença de Crohn fistulizante ativa
A eficácia foi avaliada em um estudo randomizado, duplo-cego e controlado por placebo em 94 pacientes com doença de Crohn que apresentavam fístulas por pelo menos 3 meses. Trinta e um desses pacientes foram tratados com 5 mg / kg de infliximabe. Cerca de 93% dos pacientes já haviam sido submetidos a terapia antibiótica ou imunossupressora.
O uso concomitante de dose inalterada de terapias convencionais foi permitido e 83% dos pacientes continuaram a receber pelo menos uma dessas terapias. Os pacientes receberam três doses de placebo ou infliximabe nas semanas 0, 2 e 6. O acompanhamento dos pacientes foi de 26 semanas . O desfecho primário foi o número de pacientes que apresentaram uma resposta clínica, definida como uma redução ≥ 50% da linha de base no número de fístulas purgantes após compressão leve em pelo menos dois controles consecutivos (4 semanas depois), sem aumento no uso de drogas ou cirurgia para a doença de Crohn.
68% (21/31) dos pacientes que receberam infliximabe na dose de 5 mg / kg tiveram uma resposta clínica em comparação com 26% (8/31) dos pacientes tratados com placebo (p = 0,002). O tempo médio de resposta no grupo do infliximabe foi de 2 semanas. A duração média da resposta foi de 12 semanas. Além disso, o fechamento de todas as fístulas foi observado em 55% dos pacientes que receberam infliximabe, em comparação com 13% dos pacientes que receberam placebo (p = 0,001).
Tratamento de manutenção na doença de Crohn fistulizante ativa
A eficácia de infusões repetidas de infliximabe em pacientes com doença de Crohn fistulizante foi avaliada em um estudo de 1 ano (ACCENT II). Um total de 306 pacientes receberam 3 doses de 5 mg / kg de infliximabe nas semanas 0, 2 e 6. No início do estudo , 87% dos pacientes tinham fístulas perianais, 14% tinham fístulas abdominais, 9% tinham fístulas retovaginais. O escore CDAI médio foi 180. Na semana 14, 282 pacientes foram avaliados quanto à resposta clínica e randomizados para serem tratados com placebo ou infliximabe 5 mg / kg a cada 8 semanas até a semana 46.
Os pacientes que responderam na semana 14 (195/282) foram analisados para o desfecho primário, que era o tempo entre a randomização e a perda de resposta (ver tabela 7). Corticosteroides reduzidos foram permitidos após a semana 6.
Tabela 7
Efeitos na velocidade de resposta, dados do estudo ACCENT II (pacientes que alcançaram a resposta na semana 14)
uma redução ≥ 50% da linha de base no número de fístulas de drenagem ao longo de um período de ≥ 4 semanas
b Ausência de fístulas drenantes
No início da semana 22, os pacientes que inicialmente responderam ao tratamento e que posteriormente perderam a resposta foram trocados para um retratamento ativo a cada 8 semanas com uma dose de infliximabe 5 mg / kg maior do que a dose em que haviam sido inicialmente randomizados. o grupo de manutenção com infliximabe 5 mg / kg que mudou para retratamento ativo porque perderam a resposta à redução da fístula após a semana 22, 57% (12/21) responderam ao retratamento com infliximabe 10 mg / kg a cada 8 semanas.
Não houve diferença significativa entre o placebo e o infliximabe na proporção de pacientes com fechamento sustentado de todas as fístulas até a semana 54, nos sintomas de proctalgia, abscesso e infecções do trato urinário ou no número de novas fístulas desenvolvidas durante o tratamento.
A terapia de manutenção com infliximabe a cada 8 semanas reduziu significativamente as hospitalizações relacionadas à doença e a cirurgia em comparação com o placebo. Além disso, observou-se redução do uso de corticosteroides e melhora da qualidade de vida.
Colite ulcerativa em adultos
A segurança e eficácia de Remicade foram avaliadas em dois ensaios clínicos randomizados, duplo-cegos, controlados por placebo (ACT 1 e ACT 2) em pacientes adultos com colite ulcerativa ativa moderada a grave (pontuação Mayo 6 a 12; subpontuação endoscópica ≥ 2) com resposta inadequada às terapias convencionais [corticosteroides orais, aminossalicilatos e / ou imunomoduladores (6 MP, AZA)]. A administração concomitante de doses fixas de aminossalicilatos orais, corticosteroides e / ou imunoloduladores foi permitida. Os pacientes dos estudos foram randomizados para receber placebo ou Remicade 5 mg / kg ou Remicade 10 mg / kg nas semanas 0, 2, 6, 14 e 22 e no ACT 1 nas semanas 30, 38 e 46. A redução dos corticosteroides foi permitida após 8 semanas.
Tabela 8
Efeitos na resposta clínica, remissão clínica e cicatrização da mucosa nas semanas 8 e 30.
Dados combinados de ACT 1 e 2
no p
A eficácia do Remicade na semana 54 foi avaliada no estudo ACT 1.
Na semana 54, 44,9% dos pacientes no grupo de combinação de infliximabe tiveram uma resposta clínica em comparação com 19,8% no grupo de placebo (p
Uma proporção maior de pacientes no grupo de combinação de infliximabe foi capaz de descontinuar o tratamento com corticosteroides e permanecer em remissão clínica em comparação com o grupo de placebo em ambas as semanas 30 (22,3% vs 7,2%, p
Os dados combinados dos estudos ACT 1 e ACT 2 e suas extensões, analisados desde o início até a semana 54, demonstraram uma redução nas hospitalizações relacionadas à colite ulcerativa e nas intervenções cirúrgicas após o tratamento com infliximabe. O número de hospitalizações relacionadas à colite ulcerosa foi significativamente menor nos grupos de tratamento com infliximabe 5 e 10 mg / kg em comparação com o grupo placebo (número médio de hospitalizações por 100 indivíduos por ano: 21 e 19 versus 40 no grupo placebo; p = 0,019 ep = 0,007, respectivamente).
O número de cirurgias relacionadas à colite ulcerosa também foi menor nos grupos de tratamento com infliximabe 5 e 10 mg / kg em comparação com o grupo placebo (número médio de cirurgias por 100 indivíduos por ano: 22 e 19 versus 34; p = 0,145 ep = 0,022, respectivamente).
O número de indivíduos submetidos à colectomia em qualquer momento durante as 54 semanas após a primeira infusão do agente do estudo foi coletado e combinado com os dados dos estudos ACT 1 e ACT 2 e suas extensões. Menos indivíduos foram submetidos à colectomia no infliximabe 5 mg / kg grupo (28/242 ou 11,6% [NS]) e no grupo infliximabe 10 mg / kg (18/242 ou 7,4% [p = 0,011]) em comparação com o grupo placebo (36/244; 14,8%).
A redução na incidência de colectomias também foi examinada em outro estudo duplo-cego randomizado (C0168Y06) em pacientes hospitalizados (n = 45) com colite ulcerativa ativa moderada a grave que não responderam aos corticosteroides intravenosos. E que, portanto, estavam alto risco de colectomia. Houve significativamente menos colectomias dentro de 3 meses de infusão em pacientes que receberam uma dose única de 5 mg / kg de infliximabe em comparação com pacientes que receberam placebo (29,2% versus 66,7% respectivamente, p = 0,017).
Nos estudos ACT 1 e ACT 2, o infliximabe melhorou a qualidade de vida, confirmada por uma melhora estatisticamente significativa tanto na medida de um parâmetro específico da doença, o IBDQ, quanto na melhora das 36 questões genéricas que compõem o SF-36.
Espondilite anquilosante em adultos
A eficácia e segurança do infliximabe foram estudadas em dois estudos multicêntricos duplo-cegos, controlados por placebo em pacientes com espondilite anquilosante ativa (Índice de banho de atividade da doença de espondilite anquilosante [BASDAI] pontuação ≥ 4 e dor na coluna ≥ 4 em uma escala de 1 a 10).
No primeiro estudo (P01522), que incluiu uma fase duplo-cega de 3 meses, 70 pacientes foram tratados com infliximabe 5 mg / kg ou placebo nas semanas 0, 2, 6 (35 pacientes por grupo). A partir da semana 12, os pacientes tratados até agora com placebo começaram a receber infliximabe na dose de 5 mg / kg a cada 6 semanas até a semana 54. Após o primeiro ano, 53 pacientes foram colocados em um protocolo aberto até a semana 102.
Em um segundo estudo clínico (ASSERT), 279 pacientes foram randomizados para tratamento com placebo (grupo 1, n = 78) ou infliximabe 5 mg / kg (grupo 2, n = 201) nas semanas 0, 2, 6 e a cada 6 semanas até a semana 24. Depois disso, todos os participantes do estudo continuaram com infliximabe a cada 6 semanas até a semana 96. O Grupo 1 recebeu uma dose de 5 mg / kg de infliximabe. No grupo 2, começando na semana 36, os pacientes que tiveram um BASDAI ≥ 3 por 2 visitas consecutivas foram tratados com uma dose de infliximabe de 7,5 mg / kg a cada 6 semanas até a semana 96.
No estudo ASSERT, a melhora nos sinais e sintomas foi observada a partir da semana 2. Na semana 24, o número de pacientes que tiveram uma resposta ASAS 20 foi 15/78 (19%) no grupo placebo e igual a 123/201 (61 %) no grupo de infliximabe 5 mg / kg (p
No estudo P01522, a melhora nos sinais e sintomas foi observada a partir da semana 2. Na semana 12, os pacientes que tiveram uma resposta BASDAI 50 foram 3/35 (9%) no grupo placebo e 20/35 (57%) no infliximabe 5 grupo mg / kg (p
Em ambos os estudos, a função física e a qualidade de vida, medida pelo BASFI e a pontuação do componente físico na escala SF-36, melhoraram significativamente.
Artrite psoriática em adultos
A eficácia e a segurança foram avaliadas em dois estudos multicêntricos duplo-cegos, controlados por placebo, em pacientes com artrite psoriática ativa.
No primeiro estudo clínico (IMPACT), a eficácia e segurança do infliximabe foram estudadas em 104 pacientes com artrite psoriática poliarticular ativa. Durante a fase duplo-cega de 16 semanas, os pacientes receberam 5 mg / kg de infliximabe ou palcebo. 0, 2, 6 e 14 (52 pacientes em cada grupo). A partir da semana 16, os pacientes no grupo de placebo foram trocados para infliximabe e, em seguida, todos os pacientes receberam infliximabe na dose de 5 mg / kg cada. De 8 semanas a semanas 46. Após o primeiro ano do estudo, 78 pacientes continuaram com uma extensão aberta até a semana 98.
No segundo estudo clínico (IMPACT 2), a eficácia e segurança do infliximabe foram estudadas em 200 pacientes com artrite psoriática ativa (articulações inchadas ≥ 5 e articulações doloridas ≥ 5). Quarenta e seis por cento dos pacientes continuaram com doses fixas de metotrexato ( ≤ 25 mg / semana) Durante a fase duplo-cega de 24 semanas, os pacientes receberam 5 mg / kg de infliximabe ou placebo nas semanas 0, 2, 6, 14 e 22 (100 pacientes em cada grupo). Na semana 16, 47 pacientes estavam recebendo placebo com melhora
Os principais resultados de eficácia para IMPACT e IMPACT 2 são descritos abaixo
Tabela 9
Efeitos no ACR e PASI no IMPACTO e IMPACTO 2
* Análise ITT em que os indivíduos com dados ausentes foram incluídos como Não-respondentes
na semana 98, os dados do IMPACT incluem pacientes do grupo de placebo e pacientes tratados com infliximabe que entraram na extensão de rótulo aberto
b Com base em pacientes com PASI ≥ 2,5 no início do estudo para IMPACT e pacientes com envolvimento da área de superfície corporal (BSA) psoriásico no início do estudo ≥ 3% no IMPACT 2
** Resposta PASI 75 para IMPACTO não incluída devido ao baixo N; p
No IMPACTO e IMPACTO 2, a resposta clínica foi observada já na semana 2 e foi mantida até a semana 98 e semana 54, respectivamente. A eficácia foi demonstrada com e sem o uso concomitante de metotrexato. Reduções nos parâmetros de atividade periférica característicos da artrite psoriática (como número de articulações inchadas, número de articulações doloridas / sensíveis, dactilite e presença de entesopatias) foram observadas em pacientes tratados com infliximabe.
As alterações radiográficas foram avaliadas no IMPACT2. As radiografias de mãos e pés foram coletadas no início do estudo, semana 24 e semana 54. O tratamento com infliximabe reduziu a taxa de progressão da lesão articular periférica em comparação com o tratamento com placebo no desfecho primário da semana 24, medido como alteração da linha de base na pontuação total modificada vdH -S (pontuação média ± DP foi de 0,82 ± 2,62 no grupo placebo versus - 0,70 ± 2,53 no grupo infliximabe; p
Melhoria significativa na função física avaliada pelo HAQ foi demonstrada em pacientes tratados com infliximabe.Melhorias significativas na qualidade de vida medida pelo escore resumido dos componentes físicos e mentais do SF-36 no IMPACT 2 também foram demonstradas.
Psoríase em adultos
A eficácia do infliximabe foi avaliada em dois estudos randomizados, duplo-cegos, multicêntricos, SPIRIT e EXPRESS. Os pacientes em ambos os estudos tinham psoríase em placas (BSA [Área de superfície corporal] ≥ 10% e pontuação PASI [Índice de área e gravidade da psoríase] ≥ 12 ) O endpoint primário em ambos os estudos foi a proporção de pacientes que alcançaram ≥ 75% de melhora em relação ao valor basal na pontuação PASI na semana 10.
O SPIRIT avaliou a eficácia da terapia de indução de infliximabe em 249 pacientes com psoríase em placas previamente tratados com PUVA ou terapia sistêmica. Os pacientes receberam infusões de 3 ou 5 mg / kg de infliximabe ou placebo nas semanas 0, 2 e 6. Pacientes com PGA ≥ 3 eram elegíveis para receber uma infusão adicional do mesmo tratamento na semana 26.
No SPIRIT, a proporção de pacientes que atingiram PASI 75 na semana 10 foi de 71,7% no grupo de infliximabe 3 mg / kg, 87,9% no grupo de infliximabe 5 mg / kg e no grupo de infliximabe 5 mg / kg. 5,9% no grupo grupo placebo (p 20 semanas. Nenhum fenômeno de rebote foi observado.
O EXPRESS avaliou a eficácia da terapia de indução e manutenção com infliximabe em 378 pacientes com psoríase em placas. Os pacientes receberam infusões de 5 mg / kg de infliximabe ou placebo nas semanas 0, 2 e 6, seguidos por terapia de manutenção a cada 8 semanas. Semanas até a semana 22 no grupo de placebo e até a semana 46 no grupo infliximabe. Na semana 24, o grupo placebo mudou para terapia de indução de infliximabe (5 mg / kg) seguida de terapia de manutenção com infliximabe (5 mg / kg) A psoríase ungueal foi avaliada usando o Índice de Gravidade da Psoríase Ungueal (NAPSI ) A terapia anterior com PUVA, metotrexato, ciclosporina ou acitretina foi recebida por 71,4% dos pacientes, embora não fossem necessariamente resistentes à terapia. Os resultados mais significativos são apresentados na Tabela 10. Em indivíduos tratados com infliximabe, respostas significativas ao PASI 50 foram evidente no primeiro v isita (semana 2) e respostas ao PASI 75 na segunda visita (semana 6). A eficácia no subgrupo de pacientes que já haviam se submetido a terapias sistêmicas foi semelhante à da população geral do estudo.
Tabela 10
Resumo das respostas PASI, respostas PGA e porcentagem de pacientes com todas as unhas cicatrizadas nas semanas 10, 24 e 50. EXPRESSO.
no p
b n = 292
c A análise foi realizada em indivíduos com psoríase ungueal no início do estudo (81,8% dos indivíduos). Os escores NAPSI médios no início do estudo foram 4,6 e 4,3 nos grupos de infliximabe e placebo.
Melhorias significativas da linha de base foram evidentes no DLQI (p
População pediátrica
Doença de Crohn em pacientes pediátricos (6-17 anos)
No estudo REACH, 112 pacientes (com idades entre 6 e 17 anos, idade média de 13 anos) com doença de Crohn ativa moderada a grave (CDAI pediátrico com média de 40) e com uma resposta inadequada às terapias convencionais foram tratados com 5 mg / kg infliximabe nas semanas 0, 2 e 6. Uma dose estável de 6-MP, AZA ou MTX foi necessária para todos os pacientes (35% também estavam em uso de corticosteroides no início do estudo). Os pacientes considerados pelo investigador como tendo resposta clínica na semana 10 foram então randomizados em dois grupos e receberam infliximabe 5 mg / kg a cada 8 semanas ou a cada 12 semanas como terapia de manutenção. Se a resposta foi perdida durante a manutenção, uma mudança para uma dose mais elevada (10 mg / kg) e / ou em intervalos mais curtos entre as infusões (a cada 8 semanas) foi permitida. Trinta e dois pacientes pediátricos avaliáveis para os fins do estudo foram submetidos a essa transição (9 indivíduos no grupo tratado a cada 8 semanas e 23 indivíduos no grupo tratado a cada 12 semanas). Vinte e quatro desses pacientes (75,0%) recuperaram uma resposta clínica após essa mudança.
A percentagem de doentes com resposta clínica na semana 10 foi de 88,4% (99/112). A percentagem de doentes que atingiram a remissão clínica na semana 10 foi de 58,9% (66/112).
Na semana 30, a porcentagem de pacientes em remissão clínica foi maior no grupo a cada 8 semanas (59,6%, 31/52) do que a dos pacientes no grupo de manutenção a cada 12 semanas (35,3%, 18/51; p = 0,013) . Na semana 54, os dados eram os seguintes: 55,8% (29/52) no grupo de manutenção tratado a cada 8 semanas e 23,5% (12/51) no grupo de manutenção tratado a cada 12 semanas (p
Os dados sobre fístulas foram extraídos dos escores do PCDAI. Dos 22 pacientes que tinham fístulas no início do estudo, 63,6% (14/22), 59,1% (13/22) e 68,2% (15/22) apresentaram resposta completa em relação à fístula. Nas semanas 10, 30 e 54, respectivamente, considerando em geral os grupos de manutenção tanto aqueles tratados a cada 8 semanas quanto aqueles tratados a cada 12 semanas.
Além disso, uma melhora estatística e clinicamente significativa na qualidade de vida e altura, bem como uma redução significativa no uso de corticosteroides, foi observada desde o início.
Colite ulcerosa pediátrica (6-17 anos)
A segurança e eficácia do infliximabe foram avaliadas em um ensaio clínico multicêntrico, randomizado, aberto, de grupo paralelo (C0168T72) em 60 pacientes pediátricos com idade entre 6 e 17 anos (idade média de 14,5 anos) com colite. Úlcera ativa moderada a grave ( Escore Mayo 6 a 12; subpontuação endoscópica ≥ 2) com resposta inadequada às terapias convencionais. No início do estudo, 53% dos pacientes estavam recebendo terapia imunomoduladora (6-MP, AZA e / ou MTX) e 62% dos pacientes estavam recebendo corticosteroides. imunomoduladores e redução gradual dos corticosteroides foram permitidos após a semana 0.
Todos os pacientes receberam um regime de indução de infliximabe 5 mg / kg nas semanas 0, 2 e 6. Os pacientes que não responderam ao infliximabe na semana 8 (n = 15) não receberam medicação e retornaram para um acompanhamento de avaliação de segurança. Na semana 8, 45 pacientes foram randomizados e receberam tratamento de manutenção com infliximabe 5 mg / kg a cada 8 semanas ou a cada 12 semanas.
A proporção de pacientes com resposta clínica na semana 8 foi de 73,3% (44/60). A resposta clínica na semana 8 foi semelhante entre os pacientes com e sem uso concomitante de imunomoduladores no início do estudo. A remissão clínica na semana 8 foi de 33,3% (17/51), conforme medido pelo índice de atividade da colite ulcerativa pediátrica (PUCAI).
Na semana 54, a proporção de pacientes em remissão clínica medida pela pontuação PUCAI foi de 38% (8/21) no grupo de manutenção a cada 8 semanas e 18% (4/22) no grupo de manutenção a cada 12 semanas. Semanas. Para pacientes recebendo corticosteroides no início do estudo, a proporção de pacientes em remissão e não recebendo corticosteroides na semana 54 foi de 38,5% (5/13) no grupo de manutenção a cada 8 semanas e 0% (0/13) no grupo de manutenção tratado a cada 12 semanas.
Neste estudo, havia mais pacientes na faixa etária de 12 a 17 anos do que na faixa de 6 a 11 anos (45/60 vs. 15/60). Embora o número de pacientes em cada subgrupo fosse muito pequeno para tirar conclusões firmes sobre o "efeito da idade", houve um número maior de pacientes na faixa etária mais jovem que aumentou a dose ou interrompeu o tratamento devido à eficácia inadequada.
Outras indicações pediátricas
A Agência Europeia de Medicamentos dispensou a obrigação de apresentação dos resultados dos estudos com Remicade em todos os subgrupos da população pediátrica na artrite reumatóide, artrite idiopática juvenil, artrite psoriática, espondilite anquilosante, psoríase e doença de Crohn (ver secção 4.2 para informação sobre utilização pediátrica )
05.2 "Propriedades farmacocinéticas
Infusões intravenosas únicas de 1, 3, 5, 10 ou 20 mg / kg de infliximabe aumentaram a concentração sérica máxima (Cmax) e a área sob a curva de concentração-tempo (AUC) de maneira proporcional à dose.O volume de distribuição no estado estacionário (Vd médio 3,0-4,1 litros) foi independente da dose administrada, mostrando assim que o infliximab é principalmente distribuído para o compartimento vascular. Não foi observada dependência temporal das características farmacocinéticas. A via de eliminação do infliximabe não foi caracterizada. O infliximabe não modificado não foi encontrado na urina. Não foram observadas diferenças importantes na depuração ou volume de distribuição relacionadas com a idade ou peso em doentes com artrite reumatóide. A farmacocinética do infliximab em doentes idosos não foi estudada. Não foram realizados estudos em doentes com função hepática ou hepática. Insuficiência renal .
Em doses únicas de 3, 5 ou 10 mg / kg, os valores médios de Cmax foram 77, 118 e 277 mcg / mL, respectivamente. A meia-vida terminal média com essas doses variou de 8 a 9,5 dias. Na maioria dos pacientes, na dose única recomendada de 5 mg / kg para a doença de Crohn e 3 mg / kg a cada 8 semanas para manutenção. Na artrite reumatóide, o infliximabe pode ser detectado no soro por pelo menos 8 semanas.
A administração repetida de infliximabe (5 mg / kg nas semanas 0, 2 e 6 na doença de Crohn fistulizante, 3 ou 10 mg / kg a cada 4 ou 8 semanas na artrite reumatóide) resultou em um leve acúmulo de infliximabe no soro após a segunda dose. Foi observada acumulação clinicamente relevante. Na maioria dos pacientes com doença de Crohn fistulizante, o infliximabe foi detectado no soro por 12 semanas (intervalo de 4-28 semanas) após a administração do regime.
População pediátrica
Uma "análise farmacocinética populacional com base em dados obtidos de pacientes com colite ulcerosa (N = 60), doença de Crohn (N = 112), artrite reumatóide juvenil (N = 117) e doença de Kawasaki (N = 16) com idades de 2 meses a No total, 17 anos indicaram que a exposição ao infliximabe era não linearmente dependente do peso corporal. Após a administração de 5 mg / kg de Remicade a cada 8 semanas, a exposição média esperada ao infliximabe no estado estacionário (área sob a curva concentração-tempo no estado estacionário, AUCss) em pacientes pediátricos de 6 a 17 anos de idade foi aproximadamente 20% menor do que a exposição mediana ao medicamento em estado estacionário em adultos. A AUCss mediana em pacientes pediátricos de 2 a menos de 6 anos de idade foi estimada em aproximadamente 40% mais baixa do que em adultos, embora o número de pacientes que apóiam esta estimativa seja limitado.
05.3 Dados de segurança pré-clínica
O infliximabe não apresenta reação cruzada com o TNFα em espécies animais que não humanos e chimpanzés. Portanto, os dados convencionais de segurança pré-clínica com infliximabe são limitados. encontrado, toxicidade materna, embriotoxicidade, teratogenicidade. Em um estudo de fertilidade e função reprodutiva geral, o número de camundongos grávidas foi reduzido após a administração do mesmo anticorpo análogo. Não se sabe se estes resultados foram devidos a efeitos em homens e / ou mulheres. Num estudo de toxicidade de dose repetida de 6 meses em ratos, utilizando os mesmos anticorpos análogos ao TNFα murino, foram observados depósitos cristalinos na cápsula do cristalino de alguns dos ratos machos tratados. Nenhum exame oftalmológico específico foi realizado em pacientes para avaliar a relevância deste evento em humanos. Não foram realizados estudos de longo prazo para avaliar o potencial carcinogênico do infliximabe. Em estudos realizados em camundongos com deficiência de TNFα, foi demonstrado que não há aumento de tumores quando provocados por iniciadores e / ou promotores de tumor conhecidos.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
Sacarose
Polissorbato 80
Fosfato de sódio monobásico
Fosfato de sódio dibásico
06.2 Incompatibilidade
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros medicamentos.
06.3 Período de validade
Antes da reconstituição:
3 anos a 2 ° C - 8 ° C.
Remicade pode ser armazenado em temperaturas não superiores a 25 ° C por um único período de até 6 meses, mas não além da data de validade original. A nova data de validade deve ser escrita na caixa. Depois de retirado do frigorífico, Remicade não deve ser guardado novamente no frigorífico.
Após reconstituição:
A estabilidade química e física em uso da solução reconstituída foi demonstrada por 24 horas a 25 ° C. Do ponto de vista microbiológico, o produto deve ser usado o mais rápido possível e, em qualquer caso, dentro de 3 horas após a reconstituição e diluição . Se não for usado imediatamente, os tempos de armazenamento em uso e as condições anteriores ao uso são de responsabilidade do usuário e não devem, em nenhum caso, exceder 24 horas a 2 ° C a 8 ° C.
06.4 Precauções especiais de armazenamento
Conservar no frigorífico (2 ° C - 8 ° C).
Para condições de conservação até 25 ° C antes da reconstituição do medicamento, ver secção 6.3.
Para condições de conservação após reconstituição do medicamento, ver secção 6.3.
06.5 Natureza da embalagem primária e conteúdo da embalagem
Frasco para injetáveis de vidro tipo I com rolha de borracha e friso de alumínio protegido por uma tampa de plástico, contendo 100 mg de infliximab.
Remicade está disponível em embalagens de 1, 2, 3, 4 ou 5 frascos para injectáveis. Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio
1. Calcule a dose e o número de frascos de Remicade necessários. Cada frasco para injetáveis de Remicade contém 100 mg de infliximab. Calcule o volume total necessário da solução de Remicade reconstituída.
2. Em condições assépticas, reconstitua cada frasco para injetáveis de Remicade com 10 ml de água para preparações injetáveis, utilizando uma seringa de calibre 21 (0,8 mm) ou agulha menor. Remova a aba de alumínio do frasco e limpe a tampa com um cotonete embebido em álcool 70%. Insira a agulha da seringa no frasco através do centro da rolha de borracha e direcione o fluxo de água para preparações injetáveis para a parede de vidro do frasco. Gire suavemente a solução para dissolver completamente o pó liofilizado. Não agite vigorosamente ou por muito tempo . NÃO AGITE. Pode ocorrer formação de espuma durante a reconstituição. Deixe a solução reconstituída repousar por 5 minutos. Verifique se a solução é incolor a amarela e opalescente, a solução pode ter algumas pequenas partículas translúcidas, pois o infliximabe é uma proteína. Não use a solução se você notar partículas opacas, mudança de cor ou outros corpos estranhos.
3. Diluir o volume total da dose de solução reconstituída de Remicade para 250 ml usando solução para perfusão de cloreto de sódio 9 mg / ml (0,9%). Não dilua a solução reconstituída de Remicade com qualquer outro diluente. Isto pode ser feito retirando um volume de solução de cloreto de sódio 9 mg / ml (0,9%) para perfusão do frasco de vidro ou saco de perfusão de 250 ml igual ao volume de Remicade reconstituído. Adicione lentamente o volume total da solução reconstituída de Remicade ao frasco de 250 ml ou saco de perfusão. Misture suavemente.
4. Administre a solução para perfusão durante um tempo de perfusão não inferior ao tempo de perfusão recomendado (ver secção 4.2). Use apenas um conjunto de infusão com um filtro em linha estéril, apirogênico e de baixa ligação a proteínas (diâmetro de poro de 1,2 micrômetro ou menos). Como nenhum conservante está contido, recomenda-se iniciar a administração da solução para perfusão intravenosa o mais rápido possível e dentro de 3 horas após a reconstituição e diluição. Se a reconstituição e diluição forem feitas em condições assépticas, a solução para perfusão de Remicade pode ser usada dentro de 24 horas quando armazenada entre 2 ° C e 8 ° C. A solução não utilizada não deve ser armazenada para uso posterior.
5. Não foram realizados estudos de compatibilidade física e bioquímica para avaliar a combinação de Remicade com outros agentes. Não administre Remicade concomitantemente com outros medicamentos da mesma linha intravenosa.
6. Antes da administração, inspecione visualmente Remicade para garantir que não se observam partículas ou descoloração.Se forem observadas partículas opacas, descoloração ou partículas estranhas, não utilize.
7. Medicamentos não utilizados e resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Janssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Holanda
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU / 1/99/116/001
034528012
EU / 1/99/116/002
EU / 1/99/116/003
EU / 1/99/116/004
EU / 1/99/116/005
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 13 de agosto de 1999. Data da renovação mais recente: 2 de julho de 2009.
10.0 DATA DE REVISÃO DO TEXTO
24 de setembro de 2015