Ingredientes ativos: Epoetina alfa
Binocrit 1.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Binocrit 2.000 UI / 1 mL solução injetável em seringa pré-cheia
Binocrit 3.000 UI / 0,3 mL solução injetável em seringa pré-cheia
Binocrit 4.000 UI / 0,4 mL solução injetável em seringa pré-cheia
Binocrit 5.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Binocrit 6.000 UI / 0,6 mL solução injetável em seringa pré-cheia
Binocrit 7.000 UI / 0,7 mL solução injetável em seringa pré-cheia
Binocrit 8.000 UI / 0,8 mL solução injetável em seringa pré-cheia
Binocrit 9.000 UI / 0,9 mL solução injetável em seringa pré-cheia
Binocrit 10.000 UI / 1 mL solução injetável em seringa pré-cheia
Binocrit 20.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Binocrit 30.000 UI / 0,75 mL solução injetável em seringa pré-cheia
Binocrit 40.000 UI / 1 mL solução injetável em seringa pré-cheia
Indicações Por que Binocrit é usado? Para que serve?
Binocrit contém a substância ativa epoetina alfa, uma proteína que estimula a medula óssea a produzir mais glóbulos vermelhos que transportam hemoglobina (uma substância que transporta oxigênio).
A epoetina alfa é uma cópia da proteína eritropoietina humana e atua da mesma forma.
Binocrit é usado para tratar a anemia sintomática causada por doença renal:
- em crianças em hemodiálise
- em adultos em hemodiálise ou diálise peritoneal,
- em adultos gravemente anêmicos ainda não em diálise.
Se você tem doença renal, pode ter falta de glóbulos vermelhos se o rim não produzir eritropoietina suficiente (que é necessária para a produção de glóbulos vermelhos). Binocrit é prescrito para estimular a medula óssea a produzir mais glóbulos vermelhos.
Binocrit é usado para tratar a anemia quando você está recebendo quimioterapia para tumores sólidos, linfoma maligno ou mieloma múltiplo (câncer de medula óssea) e se o seu médico decidir que você pode precisar de uma transfusão de sangue. Binocrit pode reduzir a necessidade de transfusões de sangue.
Binocrit é usado em pessoas moderadamente anêmicas que doam parte de seu sangue antes da cirurgia, para que o sangue coletado possa ser dado a elas durante ou após a cirurgia. Como o Binocrit estimula a produção de glóbulos vermelhos, os médicos podem extrair mais sangue dessas pessoas.
Binocrit é usado em adultos moderadamente anêmicos que estão prestes a fazer uma grande cirurgia ortopédica (como cirurgia de substituição de quadril ou joelho) para reduzir a necessidade potencial de transfusões de sangue.
Contra-indicações Quando Binocrit não deve ser usado
Não use Binocrit
- se tem alergia à epoetina alfa ou a qualquer outro componente deste medicamento (listados na secção 6).
- se lhe foi diagnosticado 'aplasia de glóbulos vermelhos pura (a medula óssea não consegue produzir glóbulos vermelhos suficientes) após tratamento com qualquer medicamento que estimule a produção de glóbulos vermelhos (incluindo Binocrit). Ver secção 4.
- se tem pressão arterial elevada que não é suficientemente controlada com medicamentos.
- para estimular a produção de glóbulos vermelhos (para que os médicos possam extrair mais sangue de você) se você não puder receber transfusões com seu próprio sangue durante ou após a cirurgia.
- se você está prestes a fazer uma grande cirurgia ortopédica eletiva (como cirurgia de quadril ou joelho) e:
- tem doença cardíaca severa
- tem veia ou doença arterial grave
- Tive recentemente um ataque cardíaco ou derrame
- não pode tomar medicamentos para tornar o sangue mais fluido. Binocrit pode não ser adequado para si. Discuta isso com seu médico.
Algumas pessoas precisam de medicamentos para reduzir o risco de coágulos sanguíneos durante o tratamento com Binocrit. Se não pode tomar medicamentos que previnam a formação de coágulos sanguíneos, não deve tomar Binocrit.
Precauções de uso O que você precisa saber antes de tomar Binocrit
Fale com o seu médico, farmacêutico ou enfermeiro antes de usar Binocrit.
Binocrit e outros produtos que estimulam a produção de glóbulos vermelhos podem aumentar o risco de coágulos sanguíneos em todos os pacientes. Este risco pode ser maior se você tiver outros fatores de risco para o desenvolvimento de coágulos sanguíneos (por exemplo, se você teve um coágulo sanguíneo no passado ou se está acima do peso, tem diabetes, tem doença cardíaca ou precisa deitar-se por um tempo. longo prazo devido a cirurgia ou doença). Informe o seu médico sobre qualquer situação. O seu médico o ajudará a decidir se Binocrit é adequado para você.
É importante que informe o seu médico se alguma das seguintes situações se aplicar a si.
Você pode conseguir usar Binocrit de qualquer maneira, mas primeiro precisa falar com seu médico.
Se você sabe que está com dor ou já sofreu de:
- pressão alta;
- convulsões ou ataques;
- doença hepática;
- anemia por outras causas;
- porfiria (uma doença rara do sangue).
Se for um doente com cancro, esteja ciente de que os medicamentos que estimulam a produção de glóbulos vermelhos (como o Binocrit) podem agir como fatores de crescimento e, portanto, podem teoricamente afetar a progressão do tumor.
Dependendo da sua situação pessoal, uma transfusão de sangue pode ser preferível. Discuta isso com seu médico.
Se você tem hepatite C e recebe interferon e ribavirina, você deve discutir isso com seu médico porque a combinação de epoetina alfa com interferon e ribavirina em casos raros causou uma redução no efeito do tratamento e uma condição chamada aplasia pura de glóbulos vermelhos ( PRCA), uma forma grave de anemia. Binocrit não foi aprovado para o tratamento de anemia associada à hepatite C.
Se for um doente com insuficiência renal crónica e, em particular, se não responder de forma adequada ao Binocrit, o seu médico irá verificar a dose de Binocrit que recebe porque aumentar repetidamente a dose de Binocrit se não responder ao tratamento pode aumentar o risco de problemas para o coração ou vasos sanguíneos e o risco de enfarte do miocárdio, acidente vascular cerebral e morte.
Tome especial cuidado com outros produtos que estimulam a produção de glóbulos vermelhos: Binocrit pertence a um grupo de produtos que, como a proteína humana eritropoietina, estimulam a produção de glóbulos vermelhos. O seu profissional de saúde irá sempre anotar o produto específico que está a utilizar.Se lhe for administrado qualquer medicamento deste grupo, para além do Binocrit, durante o tratamento, fale com o seu médico ou farmacêutico antes de o utilizar.
Interações Quais medicamentos ou alimentos podem alterar o efeito do Binocrit
Binocrit não costuma reagir com outros medicamentos, mas informe o seu médico se estiver a utilizar, tiver utilizado recentemente ou se vier a utilizar outros medicamentos, incluindo aqueles obtidos sem receita médica.
Se toma um medicamento denominado ciclosporina (usado, por exemplo, após transplantes renais), o seu médico pode pedir-lhe análises ao sangue para medir os seus níveis de ciclosporina enquanto estiver a tomar Binocrit.
Suplementos de ferro e outros estimulantes do sangue podem aumentar a eficácia de Binocrit.Seu médico decidirá se é certo tomá-los.
Se for a um hospital, clínica ou médico de família, diga-lhes que está a ser tratado com Binocrit. Isso pode afetar outros tratamentos ou resultados de testes.
Avisos É importante saber que:
Gravidez e amamentação
É importante informar o seu médico se alguma das seguintes situações se aplicar a si.
Você pode conseguir usar Binocrit de qualquer maneira, mas deve primeiro discutir isso com seu médico:
- se está grávida ou se suspeita de gravidez.
- se você está amamentando.
Binocrit contém sódio
Binocrit contém menos de 1 mmol (23 mg) de sódio por dose, ou seja, é praticamente “isento de sódio”.
Dose, método e tempo de administração. Como usar Binocrit: Posologia
Use este medicamento sempre de acordo com as indicações do seu médico. Em caso de dúvida, consulte seu médico.
O médico fez um exame de sangue e decidiu que você precisa do Binocrit.
Binocrit pode ser administrado por injeção:
- em uma veia ou tubo que vai para uma veia (por via intravenosa)
- ou sob a pele (por via subcutânea).
O seu médico decidirá como Binocrit será injetado. Normalmente, as injeções são administradas por um médico, enfermeiro ou outro profissional de saúde. Algumas pessoas, dependendo da razão pela qual precisam de tratamento com Binocrit, podem aprender mais tarde como injetá-lo sob a pele: consulte as instruções para a auto-injeção de Binocrit no final deste folheto informativo.
Binocrit não deve ser usado:
- após a data de validade indicada no rótulo e na embalagem externa
- se você sabe ou acredita que foi acidentalmente congelado ou
- se ocorrer uma falha no refrigerador.
A dose de Binocrit que lhe será administrada depende do seu peso corporal em quilogramas. A causa da anemia também é importante para o médico escolher a dose correta.
O seu médico irá verificar a sua pressão arterial regularmente durante a terapêutica com Binocrit.
Pessoas com doença renal
- O seu médico irá manter o seu nível de hemoglobina entre 10 e 12 g / dL, uma vez que um nível elevado de hemoglobina pode aumentar o risco de coágulos sanguíneos e morte.
- A dose inicial usual de Binocrit em adultos e crianças é de 50 Unidades Internacionais (UI) por quilograma (/ kg) de peso corporal, administrada três vezes por semana. Em pacientes submetidos a diálise peritoneal, Binocrit pode ser administrado duas vezes por semana.
- Em adultos e crianças, Binocrit é administrado por injeção numa veia (por via intravenosa) ou num tubo que vai para uma veia. Quando este acesso (através de uma veia ou tubo) não está prontamente disponível, o médico pode decidir injetar Binocrit sob a pele (por via subcutânea). Isso afeta pacientes em diálise e pacientes ainda não em diálise.
- O seu médico pedirá análises de sangue regularmente para ver como a anemia responde à terapia e pode ajustar a dose, geralmente a cada quatro semanas no máximo.
- Assim que a anemia for corrigida, o seu médico continuará a verificar o seu sangue regularmente. A posologia e frequência de administração de Binocrit podem ser posteriormente ajustadas para manter a sua resposta ao tratamento. O seu médico irá utilizar a dose eficaz mais baixa para controlar os seus sintomas. Anemia. .
- Se não responder de forma adequada a Binocrit, o seu médico irá verificar a dose que está a receber e irá informá-lo se as suas doses de Binocrit precisarem de ser alteradas.
- Se estiver a utilizar um intervalo mais longo (superior a uma vez por semana) entre as doses de Binocrit, pode não ser capaz de manter os níveis de hemoglobina adequados e pode ser necessário aumentar a dose ou frequência da administração de Binocrit.
- Você também pode receber suplementação de ferro antes e durante o tratamento com Binocrit para aumentar a eficácia do tratamento.
- Se estiver a fazer diálise quando iniciar o tratamento com Binocrit, pode ser necessário alterar o seu esquema de diálise. O médico decidirá sobre isso.
Adultos em quimioterapia
- O seu médico pode iniciar a terapia com Binocrit se a sua hemoglobina for de 10 g / dL ou menos.
- O seu médico irá manter o seu nível de hemoglobina entre 10 e 12 g / dL, pois um nível de hemoglobina elevado pode aumentar o risco de coágulos sanguíneos e morte.
- A dose inicial usual é 150 UI por quilograma de peso corporal três vezes por semana ou 450 UI por quilograma de peso corporal uma vez por semana.
- Binocrit é administrado por injeção sob a pele.
- O seu médico pedirá análises ao sangue e pode ajustar a dose dependendo de como a sua anemia responde ao tratamento.
- Você também pode receber suplementação de ferro antes e durante o tratamento com Binocrit para aumentar a eficácia do tratamento.
- O tratamento com Binocrit geralmente continuará por um mês após o final da quimioterapia.
Adultos que doam seu próprio sangue
- A dose usual é de 600 UI por quilograma de peso corporal, duas vezes por semana.
- Binocrit é administrado por injeção numa veia, imediatamente após a doação de sangue, durante 3 semanas antes da cirurgia.
- Você também pode receber suplementação de ferro antes e durante o tratamento com Binocrit para aumentar a eficácia do tratamento. Adultos agendados para cirurgia ortopédica de grande porte
- A dose recomendada é de 600 UI por quilograma de peso corporal uma vez por semana.
- Binocrit é administrado por injeção sob a pele todas as semanas, durante três semanas antes da cirurgia e no dia da cirurgia.
- Se houver necessidade de diminuir o tempo antes da cirurgia, administra-se uma dose diária de 300 UI / kg por no máximo dez dias antes da cirurgia, no dia da cirurgia e nos quatro dias seguintes.
- Se os exames de sangue mostrarem valores de hemoglobina muito altos antes da cirurgia, o tratamento será interrompido.
- Você pode receber suplementação de ferro antes e durante o tratamento com Binocrit para aumentar a eficácia do tratamento.
Instruções para injetar Binocrit sob a pele
No início do tratamento, Binocrit é geralmente injetado por uma equipe médica ou paramédica.Depois disso, o seu médico pode sugerir que você ou um cuidador aprendam a injetar Binocrit sob a pele (por via subcutânea) por conta própria.
- Não tente injetar este medicamento a si próprio a menos que o seu médico ou enfermeiro lhe tenha mostrado como.
- Sempre use Binocrit exatamente como dirigido pelo seu médico ou enfermeiro.
- Certifique-se de injetar apenas a quantidade de líquido indicada pelo seu médico ou enfermeiro.
- Use Binocrit apenas se tiver sido armazenado corretamente - consulte a seção 5, Como armazenar Binocrit.
- Antes de usar, deixe a seringa de Binocrit descansar até atingir a temperatura ambiente. Isso geralmente leva de 15 a 30 minutos. Use a seringa em até 3 dias após retirá-la da geladeira.
Retire apenas uma dose de Binocrit de cada seringa.
Se Binocrit for injetado sob a pele (por via subcutânea), o volume injetado geralmente não excede um mililitro (1 mL) por injeção única.
Binocrit é administrado isoladamente e não misturado com outros líquidos injetáveis.
Não agite as seringas Binocrit. A agitação vigorosa prolongada pode danificar o produto. Se o produto tiver sido agitado vigorosamente, não o use.
As instruções de auto-injeção de Binocrit encontram-se no final deste folheto informativo.
Se você esquecer de usar Binocrit
Dê a próxima injeção assim que se lembrar. Se faltar menos de um dia para a próxima injeção, ignore a injeção que se esqueceu e continue com o seu esquema normal. Não injete uma dose dupla.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico, enfermeiro ou farmacêutico.
Overdose O que fazer se você tiver tomado muito Binocrit
Informe imediatamente o seu médico ou enfermeiro se achar que foi injetado demasiado Binocrit. É improvável que ocorram efeitos secundários em caso de sobredosagem com Binocrit.
Efeitos colaterais Quais são os efeitos colaterais do Binocrit
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
Informe imediatamente o seu médico ou enfermeiro se notar algum dos efeitos colaterais listados.
Efeitos colaterais muito comuns
Eles podem afetar mais de 1 em cada 10 pessoas que usam Binocrit.
- Diarréia
- Náusea
- Ele vomitou
- Febre
- Congestão do trato respiratório, como nariz entupido e dor de garganta, foi relatada em pacientes com doença renal e ainda não em diálise.
Efeitos colaterais comuns
Estes podem afetar até 1 em cada 10 pessoas que usam Binocrit.
- Aumento da pressão arterial. ( um ajuste na dosagem dos medicamentos que já está a tomar para a hipertensão).
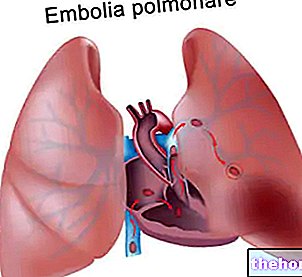
- Coágulos sanguíneos (incluindo trombose venosa profunda e embolia) que podem requerer intervenção urgente. Os sintomas podem incluir dor no peito, falta de ar e inchaço com dor e vermelhidão, geralmente nas pernas.
- Tosse.
- Erupção cutânea, que pode ser causada por uma reação alérgica.
- Dor nos ossos ou músculos.
- Sintomas semelhantes aos da gripe, como dor de cabeça, dores nas articulações, sensação de fraqueza, calafrios, cansaço e tonturas. Estes sintomas podem ser mais comuns no início do tratamento. Se sentir estes sintomas durante a injeção na veia, uma injeção mais lenta pode ajudar a evitá-los no futuro.
- Vermelhidão, ardor e dor no local da injeção.
- Inchaço nos tornozelos, pés ou dedos.
Efeitos colaterais incomuns
Estes podem afetar até 1 em 100 pessoas que usam Binocrit.
- Níveis elevados de potássio no sangue, o que pode levar a ritmos cardíacos anormais (este é um efeito colateral muito comum em pacientes em diálise).
- Convulsões.
- Congestão nasal ou das vias respiratórias.
Efeitos colaterais muito raros
Estes podem afetar até 1 em 10.000 pessoas que utilizam Binocrit.
- Sintomas da aplasia eritrocitária pura (PRCA) A aplasia eritrocitária pura (PRCA) significa que a medula óssea não produz glóbulos vermelhos suficientes. A PRCA causa "anemia súbita e grave. Os sintomas são:
- cansaço incomum,
- sentindo zonzo,
- falta de ar.
A PRCA foi relatada muito raramente, especialmente em pacientes com doença renal, após meses ou anos de tratamento com epoetina alfa e outros medicamentos que estimulam a produção de glóbulos vermelhos.
- Particularmente no início do tratamento, pode haver um aumento no número de algumas pequenas células sanguíneas (chamadas plaquetas), que normalmente estão envolvidas na formação de coágulos. O seu médico fará os exames relevantes.
Se você estiver em hemodiálise:
- Coágulos (trombose) podem se formar na fístula de diálise. É mais provável que isso aconteça se você tiver pressão arterial baixa ou se houver complicações com a fístula.
- Os coágulos também podem se formar no sistema de hemodiálise. O médico pode decidir aumentar a dose de heparina durante a diálise.
Informe imediatamente o seu médico ou enfermeiro se notar algum destes efeitos ou se notar quaisquer outros efeitos enquanto está a tomar Binocrit.
Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico, enfermeiro ou farmacêutico.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico, farmacêutico ou enfermeiro. Isto inclui quaisquer efeitos secundários possíveis não listados neste folheto. Também pode comunicar os efeitos secundários diretamente através do sistema nacional de notificação listado no Apêndice V. efeitos secundários pode ajudar fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
- Mantenha este medicamento fora da vista e do alcance das crianças.
- Não utilize este medicamento após o prazo de validade impresso no rótulo a seguir a "VAL" e na embalagem a seguir a "VAL".
- Conservar e transportar refrigerado (2 ° C - 8 ° C).
- Você pode retirar Binocrit da geladeira e guardá-lo em temperatura ambiente (até 25 ° C) por até 3 dias. Assim que a seringa for retirada do frigorífico e atingir a temperatura ambiente (até 25 ° C), deve ser utilizada no prazo de 3 dias ou rejeitada.
- Não congele nem agite.
- Conservar na embalagem original para proteger o medicamento da luz.
Não use este medicamento se notar
- que foi acidentalmente congelado ou
- que ocorreu uma falha na geladeira
- se o líquido é colorido ou se vê partículas flutuando nele
- que o selo está quebrado.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Prazo "> Outras informações
O que Binocrit contém
- O ingrediente ativo é epoetina alfa (para quantidade, ver tabela abaixo).
- Os outros componentes são di-hidrogenofosfato de sódio di-hidratado, fosfato dissódico di-hidratado, cloreto de sódio, glicina, polissorbato 80, ácido clorídrico (para ajustar o pH), hidróxido de sódio (para ajustar o pH), água para preparações injetáveis.
Qual a aparência de Binocrit e conteúdo da embalagem
Binocrit apresenta-se como uma solução injetável límpida e incolor em seringa pré-cheia. As seringas são seladas em blisters.
* Embalagens de 1, 4 ou 6 seringas pré-cheias com ou sem proteção de segurança da agulha.
Nem todos os tamanhos de embalagem podem ser comercializados.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO -
SOLUÇÃO BINOCRIT PARA INJEÇÃO EM SERINGA PRÉ-CHEIA
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA -
Binocrit 1.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 2.000 UI de epoetina alfa *, correspondendo a 16,8 mcg por mL
Uma seringa pré-cheia de 0,5 ml contém 1.000 unidades internacionais (UI), correspondendo a 8,4 mcg de epoetina alfa. *
Binocrit 2.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 2.000 UI de epoetina alfa *, correspondendo a 16,8 mcg por mL
Uma seringa pré-cheia de 1 ml contém 2.000 unidades internacionais (UI), correspondentes a 16,8 mcg de epoetina alfa. *
Binocrit 3.000 UI / 0,3 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,3 ml contém 3.000 unidades internacionais (UI), correspondentes a 25,2 mcg de epoetina alfa. *
Binocrit 4.000 UI / 0,4 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,4 ml contém 4.000 unidades internacionais (UI), correspondentes a 33,6 mcg de epoetina alfa. *
Binocrit 5.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,5 ml contém 5.000 unidades internacionais (UI), correspondendo a 42,0 mcg de epoetina alfa. *
Binocrit 6.000 UI / 0,6 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,6 ml contém 6.000 unidades internacionais (UI), correspondendo a 50,4 mcg de epoetina alfa. *
Binocrit 7.000 UI / 0,7 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,7 ml contém 7.000 unidades internacionais (UI), correspondendo a 58,8 mcg de epoetina alfa. *
Binocrit 8.000 UI / 0,8 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,8 ml contém 8.000 unidades internacionais (UI), correspondendo a 67,2 mcg de epoetina alfa. *
Binocrit 9.000 UI / 0,9 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 0,9 ml contém 9.000 unidades internacionais (UI), correspondendo a 75,6 mcg de epoetina alfa. *
Binocrit 10.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 10.000 UI de epoetina alfa *, correspondendo a 84,0 mcg por mL
Uma seringa pré-cheia de 1 ml contém 10.000 unidades internacionais (UI), correspondendo a 84,0 mcg de epoetina alfa. *
Binocrit 20.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 40.000 UI de epoetina alfa *, correspondendo a 336,0 mcg por mL
Uma seringa pré-cheia de 0,5 ml contém 20.000 unidades internacionais (UI), correspondendo a 168,0 mcg de epoetina alfa. *
Binocrit 30.000 UI / 0,75 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 40.000 UI de epoetina alfa *, correspondendo a 336,0 mcg por mL
Uma seringa pré-cheia de 0,75 ml contém 30.000 unidades internacionais (UI), correspondendo a 252,0 mcg de epoetina alfa. *
Binocrit 40.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada mL de solução contém 40.000 UI de epoetina alfa *, correspondendo a 336,0 mcg por mL
Uma seringa pré-cheia de 1 ml contém 40.000 unidades internacionais (UI), correspondendo a 336,0 mcg de epoetina alfa. *
* Produzido em células de ovário de hamster chinês (CHO) por tecnologia de DNA recombinante
Para a lista completa de excipientes, consulte a seção 6.1.
Este medicamento contém menos de 1 mmol (23 mg) de sódio por dose, ou seja, é praticamente “isento de sódio”.
03.0 FORMA FARMACÊUTICA -
Solução injetável em seringa pré-cheia (injeção)
Solução límpida e incolor
04.0 INFORMAÇÕES CLÍNICAS -
04.1 Indicações terapêuticas -
Binocrit é indicado para o tratamento da anemia sintomática associada à insuficiência renal crônica (IRC):
• em adultos e crianças em hemodiálise com 1 a 18 anos e doentes adultos a fazer diálise peritoneal (ver secção 4.4).
• em doentes adultos com insuficiência renal ainda não submetidos a diálise para o tratamento de anemia grave de origem renal acompanhada de sintomas clínicos (ver secção 4.4).
Binocrit é indicado em adultos recebendo quimioterapia para tumores sólidos, linfoma maligno ou mieloma múltiplo e em risco de transfusão, conforme indicado pelo estado geral do paciente (situação cardiovascular, anemia pré-existente no início da quimioterapia) para o tratamento de anemia e redução necessidades de transfusão.
Binocrit é indicado em adultos que fazem parte de um programa de pré-doação autólogo para aumentar a produção de sangue autólogo.O tratamento só deve ser realizado em pacientes com anemia moderada (intervalo de concentração de hemoglobina [Hb] entre 10 e 13 g / dL [6,2 e 8,1 mmol / L], sem deficiência de ferro), quando as técnicas de economia de sangue não estão disponíveis ou são insuficientes e planejadas a grande cirurgia eletiva requer uma grande quantidade de sangue (4 ou mais unidades de sangue para mulheres, 5 ou mais unidades para homens).
Binocrit é indicado em adultos sem deficiência de ferro, considerados de alto risco de complicações transfusionais, antes de grandes cirurgias ortopédicas eletivas, para reduzir a exposição a transfusões de sangue alogênico. Limitar o uso a pacientes com anemia moderada (faixa de concentração de hemoglobina entre 10 e 13 g / dL ou entre 6,2 e 8,1 mmol / L) não faz parte de um programa de pré-doação autóloga e para o qual é esperada perda moderada de sangue (900-1.800 mL).
04.2 Posologia e método de administração -
A terapia com Binocrit deve ser iniciada sob a supervisão de médicos com experiência no tratamento de pacientes com as indicações acima.
Dosagem
Todas as outras causas de anemia (deficiência de ferro, folato ou vitamina B12, intoxicação por alumínio, infecção ou inflamação, perda de sangue, hemólise e fibrose da medula óssea de qualquer origem) devem ser avaliadas e tratadas antes do início da terapia. Com epoetina alfa e ao decidir sobre um aumento da dose. Para garantir uma resposta ideal à epoetina alfa, é necessário garantir que existem reservas adequadas de ferro e, se necessário, administrar um suplemento de ferro (ver secção 4.4).
Tratamento da anemia sintomática em pacientes adultos com insuficiência renal crônica
Os sintomas e consequências da anemia podem variar de acordo com a idade, sexo e comorbidades médicas; uma avaliação individual do curso clínico e da condição de cada paciente individual é exigida pelo médico.
O intervalo de concentração de hemoglobina desejado recomendado é de 10 g / dL a 12 g / dL (6,2 a 7,5 mmol / L). Binocrit deve ser administrado de forma que os valores de hemoglobina não aumentem além de 12 g / dL (7,5 mmol / L). Deve ser evitado um aumento na hemoglobina superior a 2 g / dL (1,25 mmol / L) durante um período de semanas. Se isso ocorrer, deve ser feito o ajuste da dose apropriado.
Devido à variabilidade intra-paciente, valores individuais de hemoglobina acima e abaixo da faixa de concentração de hemoglobina desejada podem ser ocasionalmente observados: 10 g / dL (6,2 mmol / L) e 12 g / dL (7,5 mmol / L).
Níveis prolongados de hemoglobina acima de 12 g / dL (7,5 mmol / L) devem ser evitados. Se a hemoglobina aumentar em mais de 2 g / dL (1,25 mmol / L) por mês, ou se os níveis de hemoglobina forem mantidos acima de 12 g / dL (7,5 mmol / L), reduza a dose de Binocrit em 25%. Se a hemoglobina exceder 13 g / dL (8,1 mmol / L), descontinuar a terapia até que os valores caiam abaixo de 12 g / dL (7,5 mmol / L) e então retomar o tratamento com Binocrit em uma dose 25% menor do que a dose anterior.
Os pacientes devem ser monitorados de perto para garantir que a menor dose eficaz aprovada de Binocrit seja usada para o controle adequado da anemia e dos sintomas da anemia, mantendo uma concentração de hemoglobina menor ou igual a 12 g / dL (7,45 mmol / L).
Tenha cuidado ao aumentar as doses de Binocrit em doentes com insuficiência renal crónica Em doentes com resposta fraca da hemoglobina ao Binocrit, devem ser consideradas explicações alternativas para a resposta fraca (ver secções 4.4 e 5.1).
O tratamento com Binocrit consiste em duas fases: a fase de correção e a fase de manutenção.
Pacientes adultos em hemodiálise
Em pacientes em hemodiálise onde o acesso intravenoso está prontamente disponível, a administração intravenosa é preferível.
Fase de correção
A dose inicial é de 50 UI / kg, três vezes por semana.
Se necessário, aumente ou diminua a dose em 25 UI / kg (três vezes por semana) até que a faixa de concentração de hemoglobina desejada seja atingida, entre 10 g / dL e 12 g / dL (entre 6,2 e 7,5 mmol / L) (isso deve acontecer gradualmente em intervalos de pelo menos quatro semanas).
Fase de manutenção
A dose semanal total recomendada é entre 75 UI / kg e 300 UI / kg.
O ajuste de dose adequado deve ser feito para manter os valores de hemoglobina dentro do intervalo de concentração desejado de 10 g / dL a 12 g / dL (6,2 a 7,5 mmol / L).
Pacientes com valores de hemoglobina inicialmente muito baixos (8 g / dL ou> 5 mmol / L).
Pacientes adultos com insuficiência renal ainda não em diálise
Quando o acesso intravenoso não estiver disponível, Binocrit pode ser administrado por via subcutânea.
Fase de correção
Dose inicial de 50 UI / kg, 3 vezes por semana, seguida, se necessário, por incrementos de 25 UI / kg (3 vezes por semana) até que o valor desejado seja alcançado (isso deve ser feito gradualmente em intervalos de pelo menos quatro semanas )
Fase de manutenção
Durante a fase de manutenção, Binocrit pode ser administrado 3 vezes por semana ou, no caso de administração subcutânea, uma vez por semana ou uma vez a cada 2 semanas.
Devem ser feitos ajustes adequados de dose e intervalos de dosagem para manter os valores de hemoglobina no nível desejado: hemoglobina entre 10 g / dL e 12 g / dL (6,2 a 7,5 mmol / L). O prolongamento do intervalo entre as doses pode exigir um aumento na dosagem.
A dosagem máxima não deve exceder 150 UI / kg 3 vezes por semana, 240 UI / kg (até um máximo de 20.000 UI) uma vez por semana ou 480 UI / kg (até um máximo de 40.000 UI) uma vez. semanas.
Pacientes adultos em diálise peritoneal
Quando o acesso intravenoso não estiver disponível, Binocrit pode ser administrado por via subcutânea.
Fase de correção
A dose inicial é de 50 UI / kg, duas vezes por semana.
Fase de manutenção
A dose de manutenção recomendada é entre 25 UI / kg e 50 UI / kg, duas vezes por semana, em 2 injeções iguais.
O ajuste adequado da dose deve ser feito para manter os valores de hemoglobina no nível desejado, entre 10 g / dL e 12 g / dL (6,2 a 7,5 mmol / L).
Tratamento de pacientes adultos com anemia induzida por quimioterapia
Os sintomas e consequências da anemia podem variar de acordo com a idade, sexo e gravidade geral da doença; uma avaliação individual do curso clínico e condição de cada paciente individual é exigida pelo médico.
Binocrit deve ser administrado a pacientes anêmicos (por exemplo, com concentração de hemoglobina ≤ 10 g / dL (6,2 mmol / L)).
A dose inicial é de 150 UI / kg por via subcutânea, 3 vezes por semana.
Alternativamente, Binocrit pode ser administrado em uma dose inicial de 450 UI / kg por via subcutânea uma vez por semana.
O ajuste de dose adequado deve ser feito para manter os valores de hemoglobina dentro da faixa de concentração desejada de 10 g / dL a 12 g / dL (6,2 a 7,5 mmol / L).
Devido à variabilidade intra-paciente, concentrações únicas de hemoglobina acima e abaixo do intervalo de concentração de hemoglobina desejado podem ser observadas ocasionalmente. Recomenda-se que a variabilidade da hemoglobina seja abordada por meio do gerenciamento de dose ideal, levando em consideração o intervalo de concentração de hemoglobina. 10 g / dL (6,2 mmol / L) a 12 g / dL (7,5 mmol / L) desejado Concentrações de hemoglobina prolongada acima de 12 g / dL (7,5 mmol / L) devem ser evitadas; diretrizes para ajuste de dose apropriado para concentrações de hemoglobina acima de 12 g / dL (7,5 mmol) / L) são descritos abaixo.
• Se a concentração de hemoglobina aumentou pelo menos 1 g / dL (0,62 mmol / L) ou a contagem de reticulócitos aumentou? 40.000 células / mcL acima da linha de base após quatro semanas de tratamento, uma dose de 150 UI / kg três vezes por semana ou 450 UI / kg uma vez por semana deve ser mantida.
• Se o aumento na concentração de hemoglobina for
• Se o aumento na concentração de hemoglobina for
Ajuste de dose para manter uma concentração de hemoglobina entre 10 g / dL e 12 g / dL (6,2 e 7,5 mmol / L)
Se a concentração de hemoglobina aumentar em mais de 2 g / dL (1,25 mmol / L) por mês, ou se o nível de concentração de hemoglobina exceder 12 g / dL (7,5 mmol / L), reduza a dose de Binocrit em cerca de 25-50%.
Se o nível de concentração de hemoglobina exceder 13 g / dL (8,1 mmol / L), interromper a terapia até que os valores caiam abaixo de 12 g / dL (7,5 mmol / L) e, em seguida, retome o tratamento com Binocrit em uma dose 25% menor do que o dose anterior.
O regime de dosagem recomendado é mostrado na tabela a seguir:
Os pacientes devem ser monitorados de perto para garantir que a menor dose aprovada do agente estimulador da eritropoiese (agente estimulador da eritropoiese, AEE) para o controle adequado dos sintomas de anemia.
A terapia com epoetina alfa deve continuar até um mês após o final da quimioterapia.
Tratamento de pacientes cirúrgicos adultos participantes de um programa de doação autóloga
Pacientes com anemia leve (hematócrito entre 33 e 39%), que requerem pré-deposição de 4 ou mais unidades de sangue, devem ser tratados com 600 UI / kg de Binocrit por via intravenosa, duas vezes por semana, durante três semanas. Antes da cirurgia. Binocrit deve ser administrado após a conclusão do procedimento de doação.
Tratamento de pacientes adultos agendados para cirurgia ortopédica eletiva de grande porte
A dose recomendada é de 600 UI / kg de Binocrit, administrada por via subcutânea uma vez por semana durante três semanas (dias -21, -14 e -7) antes da cirurgia e no dia da cirurgia (dia 0).
Nos casos em que, por motivos médicos, seja necessário reduzir o tempo até a cirurgia para menos de três semanas, 300 UI / kg de Binocrit devem ser administrados por via subcutânea por 10 dias consecutivos antes da cirurgia, no dia da cirurgia e nos quatro dias imediatamente a seguir.
Se o nível de hemoglobina atingir ou exceder 15 g / dL (9,38 mmol / L) no período pré-operatório, Binocrit deve ser interrompido e as dosagens subsequentes não devem ser administradas.
População pediátrica
Tratamento da anemia sintomática em pacientes com insuficiência renal crônica em hemodiálise
Os sintomas e consequências da anemia podem variar de acordo com a idade, sexo e comorbidades médicas; uma avaliação individual do curso clínico e da condição de cada paciente individual é exigida pelo médico.
Em pacientes pediátricos, o intervalo de concentração de hemoglobina recomendado é de 9,5 g / dL a 11 g / dL (5,9 a 6,8 mmol / L). Binocrit deve ser administrado de forma que os valores de hemoglobina não aumentem acima de 11 g / dL (6,8 mmol / L) Deve ser evitado um aumento na hemoglobina superior a 2 g / dL (1,25 mmol / L) ao longo de um período de quatro semanas. Deve ser feito um ajuste de dose apropriado.
Os pacientes devem ser monitorados de perto para garantir que a menor dose aprovada de Binocrit seja usada para o controle adequado da anemia e dos sintomas da anemia.
O tratamento com Binocrit consiste em duas fases: a fase de correção e a fase de manutenção.
Em pacientes pediátricos em hemodiálise, onde o acesso intravenoso está prontamente disponível, a administração intravenosa é preferível.
Fase de correção
A dose inicial é de 50 UI / kg por via intravenosa, 3 vezes por semana.
Se necessário, aumente ou diminua a dose em 25 UI / kg (três vezes por semana) até que a faixa de concentração de hemoglobina desejada seja atingida, entre 9,5 g / dL e 11 g / dL (entre 5,9 e 6,8 mmol / L) ( isso deve ser feito gradualmente em intervalos de pelo menos quatro semanas).
Fase de manutenção
Deve ser feito um ajuste de dose apropriado para manter os níveis de hemoglobina dentro do intervalo de concentração desejado de 9,5 g / dL a 11 g / dL (5,9 a 6,8 mmol / L).
Normalmente, crianças com peso inferior a 30 kg precisam de doses de manutenção mais elevadas do que crianças com peso superior a 30 kg e adultos.
Pacientes pediátricos com valores basais de hemoglobina muito baixos (6,8 g / dL ou> 4,25 mmol / L).
Anemia em pacientes com insuficiência renal crônica antes de iniciar a diálise ou em diálise peritoneal
A segurança e eficácia da epoetina alfa em pacientes com insuficiência renal crônica com anemia antes do início da diálise ou em diálise peritoneal não foram estabelecidas. Os dados atualmente disponíveis para o uso subcutâneo de epoetina alfa nessas populações são descritos na seção 5.1, mas nenhuma recomendação posológica pode ser feita.
Tratamento de pacientes pediátricos com anemia induzida por quimioterapia
A segurança e eficácia da epoetina alfa em doentes pediátricos em quimioterapia não foram estabelecidas (ver secção 5.1).
Tratamento de pacientes cirúrgicos pediátricos participantes de um programa de doação autóloga
A segurança e eficácia da epoetina alfa em indivíduos pediátricos não foram estabelecidas.Não existem dados disponíveis.
Tratamento de pacientes pediátricos que aguardam cirurgia ortopédica eletiva de grande porte
A segurança e eficácia da epoetina alfa em indivíduos pediátricos não foram estabelecidas.Não existem dados disponíveis.
Método de administração
Precauções que devem ser tomadas antes de manusear ou administrar o medicamento.
Antes de usar, deixe a seringa Binocrit descansar até atingir a temperatura ambiente, o que geralmente leva de 15 a 30 minutos.
Tal como acontece com todos os outros produtos injetáveis, verifique se a solução não contém partículas e não apresenta qualquer descoloração. Binocrit é um produto estéril mas sem conservantes e destina-se a uma única utilização. Administre a quantidade necessária.
Tratamento da anemia sintomática em pacientes adultos com insuficiência renal crônica
Em pacientes com insuficiência renal crônica, onde o acesso intravenoso está regularmente disponível (pacientes em hemodiálise), a administração intravenosa de Binocrit é preferível.
Quando o acesso intravenoso não está prontamente disponível, (pacientes ainda não em diálise e pacientes em diálise peritoneal) Binocrit pode ser administrado por injeção subcutânea.
Tratamento de pacientes adultos com anemia induzida por quimioterapia
Binocrit deve ser administrado por injeção subcutânea.
Tratamento de pacientes cirúrgicos adultos participantes de um programa de doação autóloga
Binocrit deve ser administrado por via intravenosa.
Tratamento de pacientes adultos agendados para cirurgia ortopédica eletiva de grande porte
Binocrit deve ser administrado por injeção subcutânea.
Tratamento da anemia sintomática em pacientes pediátricos com insuficiência renal crônica em hemodiálise
Em pacientes pediátricos com insuficiência renal crônica, onde o acesso intravenoso está regularmente disponível (pacientes em hemodiálise), a administração intravenosa de Binocrit é preferível.
Administração intravenosa
Administre por pelo menos um a cinco minutos, dependendo da dose total. Em pacientes em hemodiálise, um bolus pode ser administrado durante a sessão de diálise por meio de um acesso venoso adequado na linha de diálise. Alternativamente, a injeção pode ser administrada no final da sessão de diálise através do tubo da agulha da fístula, seguido por 10 mL de solução salina isotônica para enxaguar o tubo e garantir que o produto seja injetado na circulação (consulte Posologia, "Pacientes adultos em hemodiálise". )
Em doentes que reagem ao tratamento com sintomas semelhantes aos da gripe, é preferível a administração mais lenta (ver secção 4.8).
Não administre Binocrit por perfusão intravenosa ou em combinação com outros medicamentos em solução (consulte a secção 6.6 para mais informações).
Administração subcutânea
Não exceda o volume máximo de 1 mL em cada local de injeção.Para injetar volumes maiores, use vários locais de injeção.
Administre a injeção nos membros ou na parede abdominal anterior.
Nos casos em que o médico determina que o paciente ou seu cuidador podem administrar Binocrit por conta própria de forma segura e eficaz, devem ser fornecidas orientações sobre a dosagem e o modo de administração corretos.
As “instruções de auto-injeção do Binocrit” encontram-se no final do Folheto Informativo.
04.3 Contra-indicações -
• Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na secção 6.1.
• Não administre Binocrit ou qualquer outra eritropoietina a doentes que desenvolvam aplasia eritrocitária pura (AEP) após tratamento com qualquer eritropoietina (ver secção 4.4).
• Hipertensão não controlada.
• Todas as contra-indicações associadas a programas de pré-doação de sangue autólogo devem ser observadas em pacientes que recebem um suplemento de Binocrit.
O uso de Binocrit em pacientes programados para cirurgia ortopédica eletiva de grande porte, que não participam de um programa de pré-doação autóloga, é contra-indicado em pacientes com doença coronariana, arterial periférica, carótida ou vascular cerebral grave, incluindo pacientes com ataque cardíaco recente acidente miocárdico ou cerebrovascular .
• Pacientes cirúrgicos que, por qualquer motivo, não podem receber profilaxia antitrombótica adequada.
04.4 Advertências especiais e precauções adequadas de uso -
Considerações gerais
Em doentes tratados com epoetina alfa, a tensão arterial deve ser cuidadosamente monitorizada e controlada conforme necessário. Epoetina alfa deve ser utilizada com precaução na presença de hipertensão não tratada, tratada de forma inadequada ou mal controlada. Pode ser necessário adicionar medicamentos. O aumentar a dose. O aumentar a dose da terapia anti-hipertensiva Descontinuar o tratamento com epoetina alfa se a pressão arterial não puder ser controlada.
Também ocorreram crises hipertensivas com encefalopatia e convulsões que requerem atenção médica imediata e cuidados intensivos durante o tratamento com epoetina alfa de doentes com tensão arterial previamente normal ou baixa, semelhante à enxaqueca, o que pode ser um sinal de alerta (ver secção 4.8).
A epoetina alfa deve ser usada com cautela em pacientes com epilepsia, história de convulsões ou condições médicas associadas a uma predisposição à atividade convulsiva, como infecções do SNC e metástases cerebrais.
A epoetina alfa deve ser utilizada com precaução em doentes com insuficiência hepática crónica.A segurança da epoetina alfa não está estabelecida em doentes com disfunção hepática.
Um “aumento da incidência de eventos trombóticos vasculares (TEV) foi observado em pacientes tratados com AEE (ver seção 4.8). Estes incluem trombose venosa e arterial e embolias (incluindo alguns casos com desfecho fatal), como trombose venosa profunda, embolia pulmonar , trombose retinal e enfarte do miocárdio Além disso, foram notificados acidentes cerebrovasculares (incluindo enfarte cerebral, hemorragia cerebral e ataques isquémicos transitórios).
O risco relatado desses TEVs deve ser cuidadosamente avaliado em relação aos benefícios esperados do tratamento com epoetina alfa, particularmente em pacientes com fatores de risco pré-existentes para TEV, incluindo obesidade e história de TEV (por exemplo, trombose venosa profunda, embolia pulmonar e acidente vascular cerebral) .
Em todos os pacientes, os níveis de hemoglobina devem ser monitorados de perto devido ao risco potencialmente maior de eventos tromboembólicos e resultado fatal se os pacientes forem tratados com níveis de hemoglobina acima do intervalo de concentração da indicação.
Durante o tratamento com epoetina alfa pode ocorrer um aumento moderado dependente da dose nas contagens de plaquetas dentro do intervalo normal. Este aumento regride durante a continuação do tratamento. Além disso, tem havido notificações de trombocitemia acima do intervalo normal. Recomenda-se. Ter as plaquetas contagem monitorada regularmente durante as primeiras oito semanas de terapia.
Todas as outras causas de anemia (deficiência de ferro, folato ou vitamina B12, intoxicação por alumínio, infecção ou inflamação, perda de sangue, hemólise e fibrose da medula óssea de qualquer origem) devem ser avaliadas e tratadas antes do início da terapia. Com epoetina alfa e ao decidir sobre um aumento da dose. Na maioria dos casos, os valores da ferritina sérica diminuem ao mesmo tempo que o hematócrito aumenta. Para garantir uma resposta ideal à epoetina alfa, é necessário garantir que existam reservas adequadas de ferro e, se necessário, administrar um suplemento de ferro (ver seção 4.2):
• Em pacientes com insuficiência renal crônica, a administração de ferro é recomendada (ferro elementar, 200 a 300 mg / dia por via oral em adultos e 100 a 200 mg / dia por via oral em pacientes pediátricos) se os níveis séricos de ferritina forem inferiores a 100 ng / mL.
• Em pacientes com câncer, a administração de ferro (ferro elementar, 200 a 300 mg / dia por via oral) é recomendada se a saturação da transferrina for inferior a 20%.
• Em pacientes que participam de um programa de pré-doação autóloga, a administração de ferro (ferro elementar, 200 mg / dia por via oral) deve ocorrer várias semanas antes do início da pré-doação autóloga, a fim de formar depósitos de ferro abundantes antes do "início da terapia com epoetina alfa, e durante toda a terapia com epoetina alfa.
• Em pacientes agendados para cirurgia ortopédica eletiva de grande porte, a administração de ferro (ferro elementar, 200 mg / dia por via oral) deve ocorrer durante toda a duração da terapia com epoetina alfa. Se possível, a administração de ferro deve ser iniciada antes do início da terapia com epoetina alfa de modo que reservas adequadas de ferro são formadas.
Muito raramente, foi observado o aparecimento ou exacerbação da porfiria em doentes tratados com epoetina alfa.Epoetina alfa deve ser utilizada com precaução em doentes com porfiria.
Para melhorar a rastreabilidade dos agentes estimuladores da eritropoiese (AEEs), o nome do agente administrado deve ser registrado (ou indicado) de forma inequívoca no prontuário do paciente.
Os pacientes devem mudar de um AEE para outro somente sob supervisão adequada.
Aplasia Eritróide Pura (PRCA)
A PRCA mediada por anticorpos foi relatada após meses ou anos de tratamento subcutâneo com epoetina, principalmente em pacientes com insuficiência renal crônica. Também foram relatados casos em pacientes com hepatite C tratados com interferon e ribavirina, na presença de terapia concomitante com AEE. A epoetina alfa não está aprovada para o tratamento da anemia associada à hepatite C.
Em pacientes nos quais há repentinamente uma falta de eficácia da terapia, definida por uma diminuição na hemoglobina (1-2 g / dL ou 0,62-1,25 mmol / L por mês) com aumento da necessidade de transfusão, deve-se determinar a contagem de reticulócitos e típica as causas de uma não resposta devem ser analisadas (deficiência de ferro, folato ou vitamina B12, intoxicação por alumínio, infecção ou inflamação, perda de sangue, hemólise e fibrose da medula óssea de qualquer origem).
No caso de diminuição paradoxal da hemoglobina e aparecimento de anemia grave associada a contagens baixas de reticulócitos, o tratamento com epoetina alfa deve ser interrompido e os anticorpos anti-eritropoietina determinados. Um exame da medula óssea também deve ser considerado para um "possível diagnóstico de AEP.
Outras terapias AEE não devem ser iniciadas devido ao risco de reação cruzada.
Tratamento da anemia sintomática em pacientes adultos e pediátricos com insuficiência renal crônica
Em pacientes com insuficiência renal crônica tratados com epoetina alfa, os níveis de hemoglobina devem ser medidos regularmente até que um nível estável seja alcançado e, posteriormente, em intervalos periódicos.
Em pacientes com insuficiência renal crônica, o aumento da hemoglobina deve ser de aproximadamente 1 g / dL (0,62 mmol / L) por mês e não deve exceder 2 g / dL (1,25 mmol / L) por mês., Para minimizar o risco de piora de hipertensão.
Em doentes com insuficiência renal crónica, a concentração de hemoglobina de manutenção não deve exceder o limite superior do intervalo de concentração de hemoglobina, conforme recomendado na secção 4.2. Observou-se um aumento do risco de morte e acontecimentos cardiovasculares graves nos ensaios clínicos. Da administração de AEE para atingir um nível de concentração de hemoglobina acima de 12 g / dL (7,5 mmol / L).
Os ensaios clínicos controlados não demonstraram nenhum benefício significativo atribuível à administração de epoetinas, uma vez que a concentração de hemoglobina excedeu os níveis necessários para controlar os sintomas de anemia e evitar transfusões de sangue.
Recomenda-se precaução ao aumentar as doses de Binocrit em doentes com insuficiência renal crónica, uma vez que doses cumulativas elevadas de epoetina podem estar associadas a um risco aumentado de mortalidade e acontecimentos cardiovasculares e cerebrovasculares graves. Em doentes com resposta insuficiente da hemoglobina às epoetinas. Explicações alternativas para a resposta insatisfatória deve ser considerada (ver seções 4.2 e 5.1).
Os doentes com insuficiência renal crónica tratados com epoetina alfa subcutânea devem ser monitorizados periodicamente quanto à perda de eficácia, definida como ausência ou redução da resposta ao tratamento com epoetina alfa em doentes que responderam anteriormente a tal terapêutica. Esta condição é caracterizada por uma diminuição prolongada da hemoglobina, apesar de um aumento na dose de epoetina alfa (ver secção 4.8).
Alguns doentes que utilizam intervalos mais longos entre as doses de epoetina alfa (mais do que uma vez por semana) podem não conseguir manter os níveis de hemoglobina adequados (ver secção 5.1) e podem necessitar de um aumento da dose de epoetina alfa. Os níveis de hemoglobina devem ser monitorados regularmente.
A trombose do shunt ocorreu em pacientes em hemodiálise, particularmente em pacientes com tendência a hipotensão ou com complicações de fístulas arteriovenosas (por exemplo, estenose, aneurismas, etc.). Nesses pacientes, a revisão precoce do shunt e uma revisão precoce do shunt são recomendadas Profilaxia antitrombótica, por exemplo com ácido acetilsalicílico.
Hipercalemia foi observada em casos isolados, embora uma ligação causal não tenha sido estabelecida. Em pacientes com insuficiência renal crônica, os eletrólitos séricos devem ser monitorados. Na presença de um nível de potássio sérico elevado ou em aumento, além do tratamento apropriado da hipercaliemia, deve-se considerar a interrupção da administração de epoetina alfa até que o nível de potássio sérico seja corrigido.
Devido ao aumento do hematócrito, um aumento da dose de heparina durante a hemodiálise é frequentemente necessário durante a terapia com epoetina alfa. Se a heparinização não for ideal, é possível que ocorra uma oclusão do sistema de diálise.
Com base nas informações atualmente disponíveis, a correção da anemia com epoetina alfa em pacientes adultos com insuficiência renal ainda não submetidos a diálise não acelera a progressão da insuficiência renal.
Tratamento de pacientes com anemia induzida por quimioterapia
Em pacientes com câncer tratados com epoetina alfa, os níveis de hemoglobina devem ser medidos regularmente até que um nível estável seja alcançado e, posteriormente, em intervalos periódicos.
As epoetinas são fatores de crescimento que estimulam principalmente a produção de eritrócitos. Os receptores de eritropoetina podem ser expressos na superfície de uma variedade de células cancerígenas. Como ocorre com todos os fatores de crescimento, existe a preocupação de que as epoetinas possam estimular o crescimento do tumor. O papel dos AEEs na progressão do tumor não pode ser descartado. Na redução da sobrevida livre de progressão. Em ensaios clínicos controlados, o uso de epoetina alfa e outros AEEs foi associado a uma redução no controle do tumor loco-regional ou uma redução na sobrevida global:
• controle locorregional reduzido em pacientes com câncer avançado de cabeça e pescoço tratados com radioterapia, quando administrado para atingir um nível de concentração de hemoglobina acima de 14 g / dL (8,7 mmol / L),
• uma redução na sobrevida global e um aumento nas mortes atribuídas à progressão do tumor em 4 meses em pacientes com câncer de mama metastático tratados com quimioterapia quando administrada para atingir uma faixa de concentração de hemoglobina de 12-14 g / dL (7,5-8,7 mmol / L) ,
• um risco aumentado de morte quando administrado para atingir um nível de concentração de hemoglobina de 12 g / dL (7,5 mmol / L) em pacientes com doenças malignas ativas, não tratados com quimioterapia ou radioterapia. O uso de AEE não é indicado nesta população de pacientes.
• um risco aumentado de 9% de progressão da doença (DP) ou morte no grupo de epoetina alfa e terapia padrão (SOC) e um risco aumentado de 15%, que não pode ser estatisticamente excluído em pacientes com câncer de mama metastático tratados com quimioterapia quando administrado para atingir uma faixa de concentração de hemoglobina de 10-12 g / dL (6,2-7,5 mmol / L).
Com base no acima exposto, em algumas condições clínicas, a transfusão de sangue deve ser o tratamento de escolha para o tratamento da anemia em pacientes com câncer. A decisão de tratar com eritropoietina recombinante deve ser baseada em uma avaliação do benefício-benefício. Risco com o envolvimento do paciente individual e deve levar em consideração o contexto clínico específico. Os fatores a serem considerados nesta avaliação devem incluir o tipo de câncer e seu estágio, o grau de anemia, a "expectativa de vida", o ambiente em que o paciente é tratado e o estado do paciente preferências (consulte a seção 5.1).
Em pacientes com câncer recebendo quimioterapia, o intervalo de 2-3 semanas entre a administração de AEE e o aparecimento de eritrócitos induzidos pela eritropoietina deve ser levado em consideração ao avaliar a adequação da terapia com epoetina alfa (pacientes em risco de transfusão).
Pacientes cirúrgicos que participam de programas de doação autóloga
Todos os avisos e precauções especiais relativos a programas de pré-doação autóloga devem ser observados; em particular, a reposição de volume deve ser realizada rotineiramente.
Pacientes agendados para cirurgia ortopédica eletiva de grande porte
Sempre siga as boas práticas de gerenciamento de sangue no período perioperatório.
Pacientes programados para cirurgia ortopédica eletiva de grande porte devem receber "profilaxia antitrombótica adequada, pois eventos trombóticos e vasculares podem ocorrer em pacientes cirúrgicos, especialmente em pacientes com doença cardiovascular subjacente. Atenção especial também deve ser dada. Cuidado em pacientes predispostos a desenvolver TVP (veia profunda Além disso, em doentes com hemoglobina basal> 13 g / dL (> 8,1 mmol / L), não pode ser excluída a possibilidade de o tratamento com epoetina alfa poder estar associado a um risco aumentado de acontecimentos trombóticos / vasculares pós-operatórios. , a epoetina alfa não deve ser utilizada em doentes com hemoglobina basal> 13 g / dL (> 8,1 mmol / L).
Excipientes
Este medicamento contém menos de 1 mmol (23 mg) de sódio por seringa pré-cheia, ou seja, é praticamente “isento de sódio”.
04.5 Interações com outros medicamentos e outras formas de interação -
Não há evidência de que o tratamento com epoetina alfa altere o metabolismo de outros medicamentos.
Os medicamentos que reduzem a eritropoiese podem reduzir a resposta à epoetina alfa.
Uma vez que a ciclosporina é ligada aos eritrócitos, existe o potencial para interação medicamentosa. Se a epoetina alfa for administrada concomitantemente com a ciclosporina, os níveis sanguíneos de ciclosporina devem ser monitorados e a dose de ciclosporina ajustada à medida que o hematócrito aumenta.
Não há evidências que indiquem uma "interação entre a epoetina alfa e o fator estimulador de colônias de granulócitos (G-CSF) ou fator estimulador de colônias de granulócitos e macrófagos (GM-CSF) em relação à diferenciação hematológica ou proliferação em amostras de biópsia tumoral. em vitro.
Em doentes adultos com cancro da mama metastático, a co-administração subcutânea de 40.000 UI / ml de epoetina alfa com 6 mg / kg de trastuzumab não teve efeito na farmacocinética do trastuzumab.
04.6 Gravidez e amamentação -
Gravidez
Não existem dados ou dados limitados sobre a utilização de epoetina alfa em mulheres grávidas Os estudos em animais revelaram toxicidade reprodutiva (ver secção 5.3).
Portanto, epoetina alfa só deve ser usada na gravidez se o benefício potencial superar o risco potencial para o feto.O uso de epoetina alfa não é recomendado em pacientes cirúrgicas grávidas que fazem parte de um programa de pré-doação autóloga.
Hora da alimentação
Não se sabe se a epoetina alfa exógena é excretada no leite humano.A epoetina alfa deve ser usada com precaução em mulheres a amamentar. Deve ser tomada uma decisão sobre a descontinuação da amamentação ou a descontinuação / abstenção da terapia com epoetina alfa, levando em consideração o benefício da amamentação para a criança e o benefício da terapia para a mulher.
O uso de epoetina alfa não é recomendado em pacientes cirúrgicos em lactação que participam de um programa de pré-doação autóloga.
Fertilidade
Não existem estudos para determinar o efeito potencial da epoetina alfa na fertilidade masculina ou feminina.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas -
Não foram realizados estudos sobre a capacidade de conduzir e utilizar máquinas Binocrit não tem ou tem uma influência negligenciável sobre a capacidade de conduzir e utilizar máquinas.
04.8 Efeitos indesejáveis -
Resumo do perfil de segurança
A reação adversa medicamentosa mais frequente durante o tratamento com epoetina alfa é o aumento da pressão arterial dependente da dose ou o agravamento da hipertensão pré-existente.A pressão arterial deve ser monitorizada, particularmente no início da terapia (ver secção 4.4).
As reações adversas medicamentosas mais frequentes observadas em estudos clínicos com epoetina alfa são as seguintes: diarreia, náuseas, vómitos, pirexia e cefaleia. Doenças semelhantes às da gripe podem ocorrer principalmente no início do tratamento.
A congestão do trato respiratório, incluindo eventos de congestão do trato respiratório superior, congestão nasal e nasofaringite, foi relatada em estudos com intervalos de dose prolongados em pacientes adultos com insuficiência renal e ainda não submetidos a diálise.
Foi observado um “aumento da incidência de acontecimentos trombóticos vasculares (TVE) em doentes tratados com AEE (ver secção 4.4).
Tabela de reações adversas
De um total de 3.262 indivíduos incluídos em 23 estudos randomizados, duplo-cegos, controlados com placebo ou tratamento padrão, o perfil de segurança geral da epoetina alfa foi avaliado em 1.992 indivíduos anêmicos. 228 indivíduos com insuficiência renal crônica tratados foram incluídos. com epoetina alfa em 4 estudos de insuficiência renal crônica (2 estudos em pré-diálise [N = 131 indivíduos expostos com insuficiência renal crônica] e 2 em diálise [N = 97 indivíduos expostos com insuficiência renal crônica]; 1.404 indivíduos com câncer expostos em 16 estudos na anemia devido à quimioterapia; 147 indivíduos expostos em 2 estudos autólogos de doação de sangue e 213 indivíduos expostos em 1 estudo perioperatório. As reações adversas medicamentosas notificadas por ≥ 1% dos indivíduos tratados com epoetina alfa nestes estudos são apresentadas na tabela abaixo.
Estimativa de frequência: muito comum (≥ 1/10); comum (≥ 1/100,
1 Identificado na experiência pós-marketing e categoria de frequência estimada com base nas taxas de relatórios espontâneos
² Comum em diálise
³ Inclui eventos arteriais e venosos, fatais e não fatais, como trombose venosa profunda, embolia pulmonar, trombose retinal, trombose arterial (incluindo infarto do miocárdio), acidentes cerebrovasculares (incluindo infarto cerebral e hemorragia cerebral), ataques isquêmicos transitórios, shunt trombose (incluindo equipamento de diálise) e trombose em aneurismas de shunt arteriovenoso
4 Discutido na subseção abaixo e / ou seção 4.4.
Descrição das reações adversas selecionadas
Foram notificadas reações de hipersensibilidade incluindo casos de erupção cutânea (incluindo urticária), reações anafiláticas e edema angioneurótico (ver secção 4.4).
Crises hipertensivas com encefalopatia e convulsões que requerem atenção médica imediata e cuidados intensivos também ocorreram durante o tratamento com epoetina alfa de pacientes com pressão arterial previamente normal ou baixa, semelhante à enxaqueca, o que pode ser um sinal de alerta (ver secção 4.4).
Aplasia eritrocitária pura mediada por anticorpos (em
População pediátrica com insuficiência renal crônica em hemodiálise
Exposição de pacientes pediátricos com insuficiência renal crônica em hemodiálise em ensaios clínicos e na experiência pós-marketing é limitado. Não foram notificadas reações adversas pediátricas específicas nesta população que não estejam mencionadas na tabela acima ou que sejam inconsistentes com a doença subjacente.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas ocorridas após a autorização do medicamento é importante porque permite a monitorização contínua da relação benefício / risco do medicamento. Os profissionais de saúde são solicitados a notificar quaisquer suspeitas de reações adversas através do sistema nacional de notificação. "Endereço www. agenziafarmaco.gov.it/it/responsabili.
04.9 Overdose -
A margem terapêutica da epoetina alfa é muito ampla. Uma sobredosagem de epoetina alfa pode causar efeitos que representam uma extensão dos efeitos farmacológicos da hormona. Na presença de níveis de hemoglobina excessivamente elevados, pode ser utilizada flebotomia. Deve recorrer a tratamentos de suporte adicionais .
05.0 PROPRIEDADES FARMACOLÓGICAS -
05.1 "Propriedades farmacodinâmicas -
Grupo farmacoterapêutico: antianêmicos, eritropoietina, código ATC: B03XA01
Binocrit é um medicamento biossimilar. Informações mais detalhadas estão disponíveis no site da Agência Europeia de Medicamentos: http://www.ema.europa.eu.
Mecanismo de ação
A eritropoietina (EPO) é um hormônio glicoproteico produzido principalmente pelo rim em resposta à hipóxia e é o regulador central da produção de eritrócitos. "A EPO está envolvida em todas as fases do desenvolvimento eritróide e seu principal efeito é expresso ao nível dos precursores eritróides. Após se ligar ao seu receptor na superfície celular, a EPO ativa as vias de transmissão de sinais que interferem na" apoptose e estimula a proliferação de células eritróides.
A EPO humana recombinante (epoetina alfa), expressa em células de ovário de hamster chinês, possui uma sequência de 165 aminoácidos idêntica à da EPO urinária humana; as duas substâncias são indistinguíveis das análises funcionais. O peso molecular aparente da eritropoietina está entre 32.000 e 40.000 daltons.
A eritropoietina é um fator de crescimento que estimula principalmente a produção de glóbulos vermelhos. Os receptores de eritropoietina podem ser expressos na superfície de diferentes tipos de células cancerígenas.
Efeitos farmacodinâmicos
Voluntários saudáveis
Após doses únicas (20.000 a 160.000 UI por via subcutânea) de epoetina alfa, foi observada uma resposta dependente da dose para os marcadores farmacodinâmicos estudados, incluindo reticulócitos, eritrócitos e hemoglobina. Um perfil de concentração-tempo definido, com pico e retorno à linha de base, foi observado para mudanças na porcentagem de reticulócitos. Um perfil menos definido foi observado para eritrócitos e hemoglobina.Em geral, todos os marcadores farmacodinâmicos aumentaram em proporção linear à dose e a resposta máxima foi alcançada em níveis de dose mais elevados.
Estudos farmacodinâmicos adicionais analisaram 40.000 UI uma vez por semana em comparação com 150 UI / kg 3 vezes por semana. Apesar das diferenças nos perfis de concentração-tempo, a resposta farmacodinâmica (medida por alterações na porcentagem de reticulócitos, hemoglobina e eritrócitos totais) foi semelhante entre esses regimes terapêuticos. Em estudos adicionais, o regime de 40.000 UI de epoetina alfa uma vez por semana foi comparado com doses variando de 80.000 a 120.000 UI por via subcutânea administradas a cada duas semanas. No geral, com base nos resultados desses estudos farmacodinâmicos em indivíduos saudáveis, o regime de 40.000 UI uma vez por semana parece ser mais eficaz em termos de produção de eritrócitos em comparação com os regimes quinzenais, embora produção de reticulócitos semelhante tenha sido observada nos regimes de uma vez por semana e a cada duas semanas.
Insuficiência renal crônica
A epoetina alfa demonstrou estimular a eritropoiese em pacientes anêmicos com insuficiência renal crônica, incluindo pacientes em diálise e pré-diálise. A primeira resposta perceptível à epoetina alfa é um aumento na contagem de reticulócitos em 10 dias, seguido por um aumento na contagem de eritrócitos, hemoglobina e hematócrito, geralmente dentro de 2 a 6 semanas. A resposta da hemoglobina varia de paciente para paciente e pode ser influenciada por depósitos de ferro e pela presença de doenças concomitantes.
Anemia induzida por quimioterapia
Epoetina alfa, administrada 3 vezes por semana ou uma vez por semana, demonstrou aumentar a hemoglobina e reduzir a necessidade de transfusões após o primeiro mês de terapia em pacientes anêmicos com câncer em quimioterapia.
Em um estudo para comparar o regime terapêutico com 150 UI / kg 3 vezes por semana e o regime terapêutico com 40.000 UI uma vez por semana em indivíduos saudáveis e em indivíduos anêmicos com câncer, os perfis temporais de mudanças na porcentagem de reticulócitos, hemoglobina e total eritrócitos foram semelhantes em ambos os regimes em indivíduos saudáveis e indivíduos com câncer anêmico. As AUCs dos respectivos parâmetros farmacodinâmicos foram semelhantes no regime de 150 UI / kg 3 vezes por semana e no regime de 40.000 UI. uma vez por semana em indivíduos saudáveis e também em sujeitos anêmicos com câncer.
Pacientes cirúrgicos adultos participando de um programa de doação autóloga
Demonstrou-se que a epoetina alfa estimula a produção de eritrócitos, permitindo o aumento da coleta de sangue autólogo e limitando a redução da hemoglobina em pacientes adultos que aguardam cirurgia eletiva de grande porte, para os quais se acredita que o pré-depósito não atende totalmente a necessidade perioperatória de sangue. Os efeitos mais evidentes são observados em pacientes com valores baixos de hemoglobina (≤ 13 g / dL).
Tratamento de pacientes adultos agendados para cirurgia ortopédica eletiva de grande porte
Em pacientes agendados para cirurgia ortopédica eletiva de grande porte com valores de hemoglobina pré-tratamento> 10 e ≤ 13 g / dL, a epoetina alfa demonstrou reduzir o risco de receber transfusões alogênicas e acelerar a recuperação eritróide (níveis aumentados de hemoglobina, níveis de hematócrito e contagens de reticulócitos).
Eficácia clínica e segurança
Insuficiência renal crônica
A epoetina alfa foi investigada em estudos clínicos em pacientes adultos anêmicos com insuficiência renal crônica, incluindo pacientes em hemodiálise e pré-diálise, para o tratamento da anemia e manutenção do hematócrito em uma faixa de concentração alvo de 30 e 36%.
Em ensaios clínicos com doses iniciais variando de 50 a 150 UI / kg três vezes por semana, aproximadamente 95% dos pacientes responderam com um aumento clinicamente significativo do hematócrito. Após aproximadamente dois meses de terapia, quase todos os pacientes eram independentes de transfusões. o hematócrito alvo foi atingido, a dose de manutenção foi definida individualmente para cada paciente.
Nos três maiores ensaios clínicos realizados em pacientes adultos em diálise, a dose de manutenção média necessária para manter o hematócrito entre 30 e 36% foi de aproximadamente 75 UI / kg administrada 3 vezes por semana.
Em um estudo duplo-cego, controlado por placebo, multicêntrico, de qualidade de vida em pacientes com insuficiência renal crônica em hemodiálise, houve uma melhora clínica e estatisticamente significativa em pacientes tratados com epoetina alfa em comparação com placebo em termos de fadiga, sintomas físicos, relacionamentos e depressão (Questionário de Doença Renal) após seis meses de terapia. Os pacientes no grupo da epoetina alfa também foram incluídos em um estudo de extensão aberto no qual as melhorias na qualidade de vida foram mostradas e mantidas por mais 12 meses.
Pacientes adultos com insuficiência renal ainda não em diálise
Em estudos clínicos conduzidos em pacientes com insuficiência renal crônica não em diálise tratados com epoetina alfa, a duração média da terapia foi de quase cinco meses. Estes pacientes responderam à terapia com epoetina alfa de forma semelhante à observada em pacientes em diálise. Em doentes com insuficiência renal crónica sem diálise, foi observado um aumento prolongado e dependente da dose do hematócrito após administração intravenosa ou subcutânea de epoetina alfa. As taxas de aumento do hematócrito foram semelhantes com ambas as vias de administração de epoetina alfa. Além disso, doses de epoetina alfa. Demonstrou-se que a epoetina alfa entre 75 e 150 UI / kg por semana mantém o hematócrito em valores entre 36 e 38% por até seis meses.
Em dois estudos com intervalos de dosagem prolongados para epoetina alfa (3 vezes por semana, uma vez por semana, uma vez a cada 2 semanas e uma vez a cada 4 semanas), alguns pacientes com intervalos de dosagem mais longos não mantiveram os níveis de hemoglobina adequados e atenderam aos critérios de retirada do protocolo para hemoglobina (0% no grupo uma vez por semana, 3,7% no grupo uma vez a cada 2 semanas e 3,3% no grupo uma vez a cada 4 semanas).
Em um estudo prospectivo randomizado, 1.432 pacientes anêmicos com insuficiência renal crônica que não foram dialisados foram avaliados. Os pacientes foram designados para tratamento com epoetina alfa para manter um nível de hemoglobina de 13,5 g / dL (acima do nível de concentração de hemoglobina recomendado) ou 11,3 g / dL. Um evento cardiovascular importante (morte, infarto do miocárdio, acidente vascular cerebral ou hospitalização por insuficiência cardíaca congestiva) ocorreu em 125 (18%) dos 715 pacientes no grupo com níveis mais elevados de hemoglobina em comparação com 97 (14%) dos 717 pacientes no com níveis mais baixos de hemoglobina (razão de risco [HR] 1,3; IC 95%: 1,0, 1,7; p = 0,03).
Foram realizadas análises post-hoc agrupadas de estudos clínicos de AEEs em pacientes com insuficiência renal crônica (em diálise, não em diálise, diabéticos e não diabéticos). Foi observada uma tendência para um aumento no risco estimado de mortalidade por todas as causas e eventos cardiovasculares e cerebrovasculares associados a doses cumulativas mais elevadas de AEE, independentemente da presença ou ausência de diabetes ou diálise (ver secção 4.2 e secção 4.4).
Tratamento de pacientes com anemia induzida por quimioterapia
A epoetina alfa foi investigada em estudos clínicos em pacientes anêmicos com câncer com tumores linfoides e sólidos e em pacientes submetidos a vários regimes de quimioterapia, incluindo platina e regimes sem platina. Nestes estudos, foi demonstrado que a epoetina alfa, administrada 3 duas vezes por semana e uma vez uma semana, aumenta a hemoglobina e reduz a necessidade de transfusões após o primeiro mês de terapia em pacientes com câncer anêmico. Em alguns estudos, a fase duplo-cega foi seguida por uma fase de rótulo aberto durante a qual todos os pacientes receberam epoetina alfa e manutenção de o efeito foi observado.
A evidência disponível indica que os doentes com doenças hematológicas malignas e tumores sólidos respondem de forma equivalente à terapêutica com epoetina alfa e que os doentes com ou sem infiltração tumoral na medula óssea respondem de forma equivalente à terapêutica com epoetina alfa. A intensidade semelhante de quimioterapia nos grupos de epoetina alfa e placebo nos estudos de quimioterapia foi demonstrada por uma "área semelhante sob a curva de neutrófilos-tempo em pacientes tratados com epoetina alfa e em pacientes tratados com placebo, bem como por uma proporção semelhante de doentes nos grupos epoetina alfa e placebo cujas contagens absolutas de neutrófilos eram inferiores a 1.000 e 500 células / mcL.
Em um estudo prospectivo, randomizado, duplo-cego e controlado por placebo conduzido com 375 pacientes anêmicos com várias doenças malignas não mielóides e tratados com quimioterapia não baseada em platina, uma redução significativa nas sequelas relacionadas à anemia (fadiga) foi observada., energia e atividade reduzida) com base nas seguintes medidas instrumentais e escalas de classificação: escala de avaliação funcional geral da terapia do câncer-anemia (FACT-An), escala de fadiga FACT-An e escala linear analógica do câncer (CLAS). dois menores, randomizados, placebo Em estudos controlados, nenhuma melhora significativa nos parâmetros de qualidade de vida foi observada nas escalas EORTC-QLQ-C30 ou CLAS, respectivamente.
A sobrevivência e a progressão do tumor foram analisadas em cinco grandes estudos controlados envolvendo um total de 2.833 pacientes, incluindo quatro estudos duplo-cegos controlados com placebo e um estudo aberto. Esses estudos envolveram pacientes em quimioterapia (dois estudos) ou populações de pacientes nas quais os AEEs não são indicados: pacientes com câncer com anemia que não fizeram quimioterapia e pacientes com câncer de cabeça e pescoço submetidos à radioterapia. Em dois estudos, o nível de concentração de hemoglobina desejado foi> 13 g / dL (8,1 mmol / L); nos estudos restantes variou de 12 a 14 g / dL (7,5 a 8,7 mmol / L). No estudo aberto, não houve diferença na sobrevida geral dos pacientes tratados com eritropoietina humana recombinante e controles.Nos quatro estudos controlados com placebo, a taxa de risco de sobrevida global variou de 1,25 a 2,47, em favor dos controles. Em comparação com os controles, esses estudos observaram um aumento estatisticamente significativo, constante e inexplicável na mortalidade em pacientes com anemia associada a várias doenças malignas comuns e tratados com eritropoietina humana recombinante. O resultado de sobrevida global não foi suficientemente explicado pelas diferenças na incidência de trombose e complicações associadas em indivíduos tratados com eritropoietina humana recombinante e em indivíduos controle.
Uma "análise de dados de pacientes individuais também foi conduzida em mais de 13.900 pacientes com câncer (recebendo quimioterapia, radioterapia, quimio-radioterapia ou não submetidos a qualquer tratamento) participando de 53 ensaios clínicos controlados envolvendo diferentes epoetinas. A meta-análise dos pacientes. Sobrevida geral os dados forneceram uma estimativa pontual da razão de risco (razão de risco) de 1,06 em favor dos controles (IC 95%: 1,00; 1,12; 53 estudos e 13.933 pacientes) e para pacientes com câncer tratados com quimioterapia a razão de risco para a sobrevida global foi 1,04 (IC 95%: 0,97, 1,11; 38 estudos e 10.441 pacientes). As meta-análises também demonstraram de forma consistente um risco relativo significativamente aumentado de eventos tromboembólicos em doentes com cancro tratados com eritropoietina humana recombinante (ver secção 4.4).
Um estudo aberto, randomizado e multicêntrico foi conduzido com 2.098 mulheres com anemia e câncer de mama metastático que receberam quimioterapia de primeira ou segunda linha. Este foi um estudo de não inferioridade desenhado para excluir um aumento de 15% no risco de progressão tumoral ou morte para epoetina alfa mais terapia padrão (SOC) em comparação com SOC sozinho. Sobrevida livre de progressão mediana (sobrevivência livre de progressão, PFS) de acordo com a avaliação do investigador da progressão da doença foi de 7,4 meses em cada braço (HR 1,09, IC 95%: 0,99, 1,20), indicando que o objetivo do estudo não foi alcançado cortar 1337 mortes foram relatadas. A sobrevida global mediana no grupo que recebeu epoetina alfa mais SOC foi de 17,2 meses, em comparação com 17,4 meses no grupo que recebeu SOC sozinho (HR 1,06, IC 95%: 0,95, 1,18). No braço que recebeu epoetina alfa mais SOC, significativamente menos pacientes receberam transfusões de eritrócitos (5,8% vs. 11,4%); no entanto, no braço que recebeu epoetina alfa mais SOC, significativamente mais pacientes (2,8% vs. 1,4%) apresentaram eventos vasculares trombóticos.
Programa de pré-doação autóloga
O efeito da epoetina alfa na facilitação da doação de sangue autólogo em pacientes com hematócrito baixo (≤ 39% na ausência de anemia ferropriva subjacente) aguardando cirurgia ortopédica de grande porte foi avaliado em um estudo duplo-cego. pacientes e um estudo simples-cego, controlado por placebo, conduzido em 55 pacientes.
No estudo duplo-cego, os pacientes foram tratados com epoetina alfa 600 UI / kg ou placebo por via intravenosa uma vez ao dia a cada 3 ou 4 dias durante 3 semanas (para um total de 6 doses). Em média, os pacientes tratados com epoetina alfa foram capazes de doar significativamente mais unidades de sangue para o pré-depósito (4,5 unidades) em comparação com os pacientes tratados com placebo (3,0 unidades).
No estudo cego, os pacientes foram tratados com epoetina alfa 300 UI / kg ou 600 UI / kg ou placebo por via intravenosa uma vez ao dia a cada 3 ou 4 dias durante 3 semanas (para um total de 6 doses). Estes pacientes tratados com epoetina alfa também foram capazes de doar significativamente mais unidades de sangue ao pré-depósito (epoetina alfa 300 UI / kg = 4,4 unidades; epoetina alfa 600 UI / kg = 4,7 unidades) em comparação com pacientes tratados com placebo (2,9 unidades) )
A terapia com epoetina alfa reduziu o risco de exposição ao sangue alogênico em 50% em comparação com pacientes que não receberam epoetina alfa.
Cirurgia ortopédica eletiva principal
O efeito da epoetina alfa (300 UI / kg ou 100 UI / kg) na exposição a transfusões de sangue alogênico foi avaliado em um estudo clínico duplo-cego controlado por placebo em pacientes adultos não ironopênicos aguardando uma cirurgia ortopédica principal eletiva em o quadril ou joelho. A epoetina alfa foi administrada por via subcutânea nos 10 dias anteriores à cirurgia, no dia da cirurgia e durante quatro dias após a cirurgia. Os pacientes foram estratificados pela hemoglobina basal (≤10 g / dL,> 10 a ≤13 g / dL e> 13 g / dL).
Epoetina alfa 300 UI / kg reduziu significativamente o risco de transfusão alogênica em pacientes com hemoglobina pré-tratamento variando de> 10 a ≤13 g / dL. Dezesseis por cento dos pacientes tratados com epoetina alfa 300 UI / kg, 23% dos pacientes tratados com epoetina alfa 100 UI / kg e 45% dos pacientes tratados com placebo necessitaram de transfusões.
Em um estudo aberto de grupo paralelo em indivíduos adultos sem deficiência de ferro com hemoglobina pré-tratamento variando de ≥10 a ≤13 g / dL aguardando cirurgia ortopédica de quadril ou joelho, a epoetina alfa foi comparada. 300 UI / kg por dia por via subcutânea nos 10 dias anteriores à cirurgia, no dia da cirurgia e nos quatro dias após a cirurgia com epoetina alfa 600 UI / kg por via subcutânea uma vez por semana durante as 3 semanas anteriores à cirurgia. intervenção e no dia da intervenção.
Da fase de pré-tratamento para a fase pré-operatória, o aumento médio da hemoglobina no grupo de 600 UI / kg por semana (1,44 g / dL) foi o dobro do grupo de 300 UI / dL. Kg por dia (0,73 g / dL). Os níveis médios de hemoglobina foram semelhantes nos dois grupos de tratamento ao longo do período pós-operatório.
A resposta eritropoiética observada em ambos os grupos de tratamento resultou em taxas de transfusão semelhantes (16% no grupo de 600 UI / kg por semana e 20% no grupo de 300 UI / kg por dia).
População pediátrica
Insuficiência renal crônica
A epoetina alfa foi avaliada em um estudo clínico aberto, não randomizado, de intervalo aberto, de 52 semanas em pacientes pediátricos com insuficiência renal crônica em hemodiálise. A mediana de idade dos pacientes incluídos no estudo foi de 11,6 anos (variação de 0,5 a 20,1 anos).
A epoetina alfa foi administrada em doses de 75 UI / kg / semana por via intravenosa, dividida em 2 ou 3 doses após a diálise, titulada para 75 UI / kg / semana em intervalos de 4 semanas (até um máximo de 300 UI / kg / semana) para obter um aumento da hemoglobina de 1 g / dL / mês. A faixa de concentração de hemoglobina desejada era de 9,6 a 11,2 g / dL. Oitenta e um por cento dos pacientes alcançaram esse nível de concentração de hemoglobina. O tempo médio para atingir a meta foi de 11 semanas e a dose média para a meta foi de 150 UI / kg / semana. Dos pacientes que atingiram a meta, 90% o alcançaram no regime de 3 vezes por semana.
Após 52 semanas, 57% dos pacientes permaneceram no estudo, recebendo uma dose mediana de 200 UI / kg / semana.
Os dados clínicos relativos à administração subcutânea em crianças são limitados. Em 5 estudos abertos não controlados com um pequeno número de pacientes (o número de pacientes variou de 9-22, para um total de N = 72), a epoetina alfa foi administrada por via subcutânea a crianças com uma dose única a partir de 100 UI / kg / semana para 150 UI / kg / semana, podendo aumentar até 300 UI / kg / semana. Nestes estudos, a maioria dos pacientes foi pré-dialisada (N = 44), 27 pacientes estavam em diálise peritoneal e dois faziam hemodiálise e a idade dos pacientes variou de 4 meses a 17 anos. Em geral, esses estudos têm limitações metodológicas, mas o tratamento foi associado a tendências positivas para níveis mais elevados de hemoglobina. Não foram relatados eventos adversos inesperados (ver seção 4.2).
Anemia induzida por quimioterapia
Epoetina alfa 600 UI / kg (administrada por via intravenosa ou subcutânea uma vez por semana) foi avaliada em um estudo randomizado, duplo-cego, controlado por placebo de 16 semanas e em um estudo randomizado, controlado, aberto e com duração de 20 semanas em pacientes pediátricos com anemia submetida a quimioterapia mielossupressora para o tratamento de várias doenças malignas não mielóides na infância.
No estudo de 16 semanas (n = 222), em pacientes tratados com epoetina alfa, não houve efeito significativo na qualidade de vida em pacientes pediátricos relatado pelos próprios pacientes ou por seus pais ou nas pontuações do Módulo do Câncer em comparação com o placebo ( Além disso, não houve diferença estatística entre a porcentagem de pacientes que necessitaram de transfusões de glóbulos vermelhos no grupo que recebeu epoetina alfa e aquele que recebeu placebo.
No estudo de 20 semanas (n = 225), não houve diferença significativa no endpoint primário de eficácia, ou seja, na proporção de pacientes que necessitaram de transfusão de glóbulos vermelhos após o dia 28 (62% dos pacientes que receberam epoetina alfa vs. 69 % de pacientes recebendo terapia padrão).
05.2 "Propriedades farmacocinéticas -
Absorção
Após a injeção subcutânea, os níveis séricos de epoetina alfa atingiram o pico entre 12 e 18 horas após a administração. Não houve acumulação após a administração de doses múltiplas de 600 UI / kg por via subcutânea uma vez por semana.
A biodisponibilidade absoluta da injeção subcutânea de epoetina alfa é de aproximadamente 20% em indivíduos saudáveis.
Distribuição
O volume médio de distribuição foi de 49,3 mL / kg após doses intravenosas de 50 e 100 UI / kg em indivíduos saudáveis. Após a administração intravenosa de epoetina alfa em indivíduos com insuficiência renal crônica, o volume de distribuição variou de 57 a 107 mL / kg após doses únicas (12 UI / kg) e de 42 a 64 mL / kg após doses múltiplas, respectivamente. (48 -192 UI / kg). Portanto, o volume de distribuição é ligeiramente maior do que o espaço plasmático.
Eliminação
A meia-vida da epoetina alfa após administração intravenosa de doses múltiplas é de aproximadamente 4 horas em indivíduos saudáveis.
A meia-vida após a administração subcutânea é estimada em aproximadamente 24 horas em indivíduos saudáveis.
A CL / F média para os regimes de 150 UI / kg 3 vezes por semana e 40.000 UI uma vez por semana em indivíduos saudáveis foram 31,2 e 12,6 mL / h / kg, respectivamente. A CL / F média para os regimes de 150 UI / kg 3 vezes por semana e 40.000 UI uma vez por semana em indivíduos com câncer anêmico foi de 45,8 e 11,3 mL / h / kg, respectivamente. Na maioria dos indivíduos com câncer anêmico submetidos à quimioterapia cíclica, CL / F foi menor após doses subcutâneas de 40.000 UI uma vez por semana e 150 UI / kg 3 vezes por semana em comparação com os valores observados em indivíduos saudáveis.
Linearidade / Não linearidade
Em indivíduos saudáveis, foi observado um aumento proporcional à dose nas concentrações séricas de epoetina alfa após administração intravenosa de 150 e 300 UI / kg 3 vezes por semana. A administração de doses únicas entre 300 e 2.400 UI / kg de epoetina alfa por via subcutânea resultou numa correlação linear entre a Cmax média e a dose e entre a AUC e a dose média.Uma correlação inversa entre a depuração aparente e a dose foi observada em indivíduos saudáveis.
Em estudos para examinar o aumento do intervalo de dosagem (40.000 IU uma vez por semana e 80.000, 100.000 e 120.000 IU a cada duas semanas), uma correlação linear, mas não proporcional à dose, foi observada entre a Cmax média e a dose e entre a AUC média e dose no estado estacionário.
Relações farmacocinéticas / farmacodinâmicas
A epoetina alfa exibe um efeito dose-dependente nos parâmetros hematológicos que é independente da via de administração.
População pediátrica
Após a administração intravenosa de doses múltiplas de epoetina alfa, observou-se uma meia-vida de aproximadamente 6,2 a 8,7 horas em indivíduos pediátricos com insuficiência renal crônica.O perfil farmacocinético da epoetina alfa em crianças e adolescentes parece semelhante ao dos adultos.
Os dados farmacocinéticos em neonatos são limitados.
Um estudo em 7 bebês prematuros com muito baixo peso ao nascer e 10 adultos saudáveis que receberam eritropoietina i.v. sugeriram que o volume de distribuição era aproximadamente 1,5 a 2 vezes maior em bebês prematuros do que em adultos saudáveis e que a depuração era aproximadamente 3 vezes maior em bebês prematuros do que em adultos saudáveis.
Insuficiência renal
Em pacientes com insuficiência renal crônica, a meia-vida da epoetina alfa administrada por via intravenosa é ligeiramente mais longa, aproximadamente 5 horas, em comparação com indivíduos saudáveis.
05.3 Dados de segurança pré-clínica -
Em estudos de toxicologia de dose repetida em cães e ratos, mas não em macacos, a terapia com epoetina alfa foi associada a fibrose subclínica da medula óssea. A fibrose da medula óssea é uma complicação conhecida da insuficiência renal crônica em humanos e pode estar relacionada a hiperparatireoidismo secundário ou fatores desconhecidos. A incidência de fibrose da medula óssea não aumentou em um estudo realizado com pacientes em hemodiálise tratados por 3 anos com epoetina alfa em comparação com um grupo controle compatível de pacientes em diálise não tratados com epoetina alfa.
A epoetina alfa não induz mutação genética em bactérias (teste de Ames), aberrações cromossômicas em células de mamíferos, micronúcleos em camundongos ou mutação genética no locus HGPRT.
Não foram realizados estudos de carcinogenicidade de longo prazo. Dados discordantes existentes na literatura, com base nos resultados obtidos em vitro com amostras de tumor humano, sugere um possível papel das eritropoietinas na proliferação do tumor. O significado clínico é incerto.
Em culturas de células da medula óssea humana, a epoetina alfa estimula especificamente a eritropoiese e não afeta a leucopoiese. Não houve efeito citotóxico da epoetina alfa nas células da medula óssea.
Em estudos em animais, a epoetina alfa mostrou induzir uma redução no peso corporal fetal, um atraso na ossificação e um aumento na mortalidade fetal quando administrada em doses semanais aproximadamente 20 vezes a dose semanal recomendada em humanos. São consideradas secundárias à da mãe. o ganho de peso corporal reduzido e seu significado para os humanos são desconhecidos em níveis de dose terapêutica.
06.0 INFORMAÇÕES FARMACÊUTICAS -
06.1 Excipientes -
Fosfato monobásico de sódio dihidratado
Fosfato dissódico dihidratado
Cloreto de Sódio
Glicina
Polissorbato 80
Água para injetáveis
Ácido clorídrico (para ajustar o pH)
Hidróxido de sódio (para ajustar o pH)
06.2 Incompatibilidade "-
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros medicamentos.
06.3 Período de validade "-
2 anos
06.4 Precauções especiais de armazenamento -
Conservar e transportar refrigerado (2 ° C - 8 ° C). Esta faixa de temperatura deve ser rigorosamente observada até a administração ao paciente.
Para uso em ambulatório, o medicamento pode ser retirado do frigorífico, sem o devolver, por um período máximo de 3 dias a uma temperatura não superior a 25 ° C. Se o medicamento não tiver sido utilizado no final deste período, deve ser descartado.
Não congele nem agite.
Conservar na embalagem original para proteger o medicamento da luz.
06.5 Natureza da embalagem primária e conteúdo da embalagem -
Seringas pré-cheias (vidro tipo I), com ou sem dispositivo de segurança da agulha, com rolha êmbolo (borracha de Teflon) seladas em embalagens de blister.
Binocrit 1.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,5 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 2.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 1 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 3.000 UI / 0,3 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,3 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 4.000 UI / 0,4 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,4 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 5.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,5 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 6.000 UI / 0,6 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,6 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 7.000 UI / 0,7 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,7 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 8.000 UI / 0,8 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,8 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 9.000 UI / 0,9 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,9 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 10.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 1 ml de solução injetável.
Embalagens de 1 ou 6 seringas.
Binocrit 20.000 UI / 0,5 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,5 ml de solução injetável.
Embalagens de 1, 4 ou 6 seringas.
Binocrit 30.000 UI / 0,75 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 0,75 ml de solução injetável.
Embalagens de 1, 4 ou 6 seringas.
Binocrit 40.000 UI / 1 mL solução injetável em seringa pré-cheia
Cada seringa pré-cheia contém 1 ml de solução injetável.
Embalagens de 1, 4 ou 6 seringas.
Nem todos os tamanhos de embalagem podem ser comercializados.
06.6 Instruções de uso e manuseio -
Binocrit não deve ser usado e deve ser descartado
• se o líquido é colorido ou se você vê partículas flutuando nele
• se o selo estiver quebrado
• se for conhecido ou assumido que pode ter sido acidentalmente congelado ou
• se ocorreu uma falha no refrigerador
As seringas pré-cheias estão prontas a usar (ver secção 4.2). A seringa pré-cheia não deve ser agitada. As seringas estão marcadas com graduações em relevo; isto permite a utilização parcial, se necessário. Cada graduação corresponde a um volume de 0, 1 mL. O produto é para uso único. Retire apenas uma dose de Binocrit de cada seringa e descarte qualquer solução desnecessária antes de injetar.
Usando a seringa pré-cheia com proteção de segurança da agulha
A proteção de segurança da agulha cobre a agulha após a injeção e evita que o operador se machuque. O dispositivo não interfere com o uso normal da seringa. Empurre lenta e uniformemente o êmbolo até que toda a dose seja liberada e o êmbolo não possa ser pressionado mais. Enquanto continua a empurrar o êmbolo, afaste a seringa do paciente. O dispositivo de segurança cobre a agulha assim que o êmbolo é liberado.
Usando a seringa pré-cheia sem proteção de segurança da agulha
Administre a dose de acordo com o procedimento padrão.
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA "AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO" -
Sandoz GmbH
Biochemiestr. 10
A-6250 Kundl
Áustria
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO -
Binocrit 1.000 UI / 0,5 mL solução injetável em seringa pré-cheia
EU / 1/07/410/001 - 1000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,5 ml (2000 UI / ml) 1 seringa pré-cheia - AIC n. 038190017 / E
EU / 1/07/410/002 - 1000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) por via subcutânea ou intravenosa 0,5 ml (2000 UI / ml) 6 seringas pré-cheias - AIC n. 038190029 / E
EU / 1/07/410/027 - 1000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,5 ml (2000 UI / ml) 1 seringa pré-cheia de 0,5 ml com dispositivo de segurança para agulha - AIC n. 038190272 / E
EU / 1/07/410/028 - 1000UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 0,5 ml (2000 UI / ml) - 6 seringas pré-cheias de 0,5 ml com agulha do dispositivo segurança - AIC N. 038190284 / E
Binocrit 2.000 UI / 1 mL solução injetável em seringa pré-cheia
EU / 1/07/410/003 - 2000 UI / 1,0 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 1,0 ml (2000 UI / ml) 1 seringa pré-cheia - AIC n. 038190031 / E
EU / 1/07/410/004 - 2000 UI / 1,0 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 1,0 ml (2000 UI / ml) 6 seringas pré-cheias - AIC n. 038190043 / E
EU / 1/07/410/029 - 2000IU / 1ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 1ml (2000IU / ml) 1 seringa pré-cheia de 1ml com dispositivo de segurança de agulha - AIC N. 038190296 / E
EU / 1/07/410 / 030-2000IU / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 1 ml (2000 UI / ml) - 6 seringas pré-cheias de 1 ml com segurança de agulha dispositivo - AIC N. 038190308 / E
Binocrit 3.000 UI / 0,3 mL solução injetável em seringa pré-cheia
EU / 1/07/410/005 - 3000 UI / 0,3 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,3 ml (10000 UI / ml) 1 seringa pré-cheia - AIC n. 038190056 / E
EU / 1/07/410/006 - 3000 UI / 0,3 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,3 ml (10000 UI / ml) 6 seringas pré-cheias - AIC n. 038190068 / E
EU / 1/07/410 / 031- 3000IU / 0,3ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,3ml (10000IU / ml) 1 seringa pré-cheia de 0,3ml com dispositivo de segurança por agulha - AIC N. 038190310 / E
EU / 1/07/410/032 - 3000IU / 0,3ml solução injetável em seringa pré-cheia (vidro) - uso intravenoso subcutâneo - 0,3ml (10000 UI / ml) - 6 seringas pré-cheias de 0,3ml com agulha de segurança - AIC N. 038190322 / E
Binocrit 4.000 UI / 0,4 mL solução injetável em seringa pré-cheia
EU / 1/07/410/007 - 4000 UI / 0,4 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,4 ml (10000 UI / ml) 1 seringa pré-cheia - AIC n. 038190070 / E
EU / 1/07/410/008 - 4000 UI / 0,4 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,4 ml (10000 UI / ml) 6 seringas pré-cheias - AIC n. 038190082 / E
EU / 1/07/410/033 - 4000IU / 0,4ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,4ml (10000IU / ml) 1 seringa pré-cheia de 0,4ml com dispositivo de segurança por agulha - AIC N. 038190334 / E
EU / 1/07/410/034 - 4000IU / 0,4 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 0,4 ml (10000 UI / ml) - 6 seringas pré-cheias de 0,4 ml com segurança para o dispositivo agulha - AIC N. 038190346 / E
Binocrit 5.000 UI / 0,5 mL solução injetável em seringa pré-cheia
EU / 1/07/410/009 - 5000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,5 ml (10000 UI / ml) 1 seringa pré-cheia - AIC n. 038190094 / E
EU / 1/07/410/010 - 5000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,5 ml (10000 UI / ml) 6 seringas pré-cheias - AIC n. 038190106 / E
EU / 1/07/410/035 - 5000IU / 0,5ml solução injetável em seringa pré-cheia (vidro) - administração intravenosa subcutânea - 0,5ml (10000 UI / ml) - 1 seringa pré-cheia 0,5ml com agulha de segurança para dispositivo - AIC N. 038190359 / E
EU / 1/07/410/036 - 5000IU / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea intravenosa - 0,5 ml (10000 UI / ml) - 6 seringas pré-cheias de 0,5 ml com segurança para o dispositivo para agulha -AIC N. 038190361 / E
Binocrit 6.000 UI / 0,6 mL solução injetável em seringa pré-cheia
EU / 1/07/410/011 - 6000 UI / 0,6 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,6 ml (10.000 UI / ml) 1 seringa pré-cheia - AIC n . 038190118 / E
EU / 1/07/410/012 - 6000 UI / 0,6 ml solução injetável em seringa pré-cheia (vidro) por via subcutânea ou intravenosa 0,6 ml (10.000 UI / ml) 6 seringas pré-cheias - AIC n . 038190120 / E
EU / 1/07/410/037 - 6000IU / 0,6 ml solução injetável em seringa pré-cheia (vidro) - administração intravenosa subcutânea - 0,6 ml (10000 UI / ml) - 1 seringa pré-cheia de 0,6 ml com segurança para o dispositivo agulha - AIC N. 038190373 / E
EU / 1/07/410/038 - 6000IU / 0,6 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 0,6 ml (10.000 UI / ml) - 6 seringas pré-cheias de 0 , 6 ml com agulha de segurança para dispositivo - AIC N. 038190385 / E
Binocrit 7.000 UI / 0,7 mL solução injetável em seringa pré-cheia
EU / 1/07/410/017 - 7000IU / 0,7 ml solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 0,7 ml (10000IU / ml) 1 seringa pré-cheia - AIC No . 038190171 / AND
EU / 1/07/410/018 - 7000IU / 0,7 ml solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 0,7 ml (10000IU / ml) 6 seringas pré-cheias - AIC No . 038190183 / AND
EU / 1/07/410/039 - 7000IU / 0,7ml solução injetável em seringa pré-cheia (vidro) - uso subcutâneo ou intravenoso - 0,7ml (10000IU / ml) 1 seringa pré-cheia de 0,7ml com dispositivo de segurança por agulha - AIC N. 038190397 / E
EU / 1/07/410/040 - 7000IU / 0,7 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,7 ml (10000IU / ml) 6 seringas pré-cheias de 0,7 ml com dispositivo de segurança por agulha - AIC N. 038190409 / E
Binocrit 8.000 UI / 0,8 mL solução injetável em seringa pré-cheia
EU / 1/07/410/013 - 8000 UI / 0,8 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,8 ml (10.000 UI / ml) 1 seringa pré-cheia - AIC n . 038190132 / E
EU / 1/07/410/014 - 8000 UI / 0,8 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 0,8 ml (10.000 UI / ml) 6 seringas pré-cheias - AIC n . 038190144 / E
EU / 1/07/410/041 - 8000IU / 0,8 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 0,8 ml (10000 UI / ml) - 1 seringa pré-cheia de 0,8 ml com segurança para o dispositivo agulha - AIC N. 038190411 / E
EU / 1/07/410/042 - 8000IU / 0,8 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 0,8 ml (10000 UI / ml) - 6 seringas pré-cheias de 0,8 ml com segurança para o dispositivo agulha - AIC N. 038190423 / E
Binocrit 9.000 UI / 0,9 mL solução injetável em seringa pré-cheia
EU / 1/07/410/019 - 9000IU / 0,9 ml solução injetável em seringa pré-cheia - seringa pré-cheia para uso subcutâneo ou intravenoso (vidro) 0,9 ml (10000IU / ml) 1 seringa pré-cheia pré-cheia - AIC No. 038190195 / AND
EU / 1/07/410/020 - 9000UI / 0,9 ml solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 0,9 ml (10000UI / ml) 6 seringas pré-cheias - AIC No . 038190207 / AND
EU / 1/07/410/043 - 9000IU / 0,9 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,9 ml (10000IU / ml) 1 seringa pré-cheia de 0,9 ml com dispositivo de segurança por agulha - AIC N. 038190435 / E
EU / 1/07/410/044 - 9000IU / 0,9 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,9 ml (10000IU / ml) 6 seringas pré-cheias de 0,9 ml com dispositivo de segurança por agulha - AIC N. 038190447 / E
Binocrit 10.000 UI / 1 mL solução injetável em seringa pré-cheia
EU / 1/07/410/015 - 10000 UI / 1,0 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 1,0 ml (10000 UI / ml) 1 seringa pré-cheia - AIC n. 038190157 / E
EU / 1/07/410/016 - 10000 UI / 1,0 ml solução injetável em seringa pré-cheia (vidro) via subcutânea ou intravenosa 1,0 ml (10000 UI / ml) 6 seringas pré-cheias - AIC n. 038190169 / E
EU / 1/07/410/045 - 10000IU / 1 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 1 ml (10000 UI / ml) - 1 seringa pré-cheia de 1 ml com dispositivo de segurança para agulha - AIC N. 038190450 / E
EU / 1/07/410/046 - 10000IU / 1 ml solução injetável em seringa pré-cheia (vidro) - via intravenosa subcutânea - 1 ml (10000 UI / ml) - 6 seringas pré-cheias de 1 ml com dispositivo de segurança para agulha - AIC N. 038190462 / E
Binocrit 20.000 UI / 0,5 mL solução injetável em seringa pré-cheia
EU / 1/07/410/021 - 20.000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa 0,5 ml (40.000 UI / ml) 1 seringa pré-cheia - AIC n. 038190219 / E
EU / 1/07/410/022 - 20.000 UI / 0,5 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa 0,5 ml (40.000 UI / ml) 6 seringas pré-cheias - AIC n. 038190221 / E
EU / 1/07/410/047 - 20000IU / 0,5ml solução injetável em seringa pré-cheia (vidro) - uso subcutâneo ou intravenoso - 0,5ml (40000IU / ml) 1 seringa pré-cheia de 0,5ml com dispositivo de segurança por agulha - AIC N. 038190474 / E
EU / 1/07/410/053 - 20.000 UI / 0,5 ML - solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 0,5 ML (40.000 UI / ML) - 4 pré-cheia seringas com dispositivo de segurança para agulha AIC: 038190536 / E
EU / 1/07/410/048 - 20000IU / 0,5ml solução injetável em seringa pré-cheia (vidro) - uso subcutâneo ou intravenoso - 0,5ml (40000IU / ml) 6 seringas pré-cheias de 0,5ml com dispositivo de segurança por agulha - AIC N. 038190486 / E
Binocrit 30.000 UI / 0,75 mL solução injetável em seringa pré-cheia
EU / 1/07/410/023 - 30.000 UI / 0,75 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa 0,75 ml (40.000 UI / ml) 1 seringa pré-cheia - AIC n. 038190233 / E
EU / 1/07/410/024 - 30.000 UI / 0,75 ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa 0,75 ml (40.000 UI / ml) 6 seringas pré-cheias - AIC n. 038190245 / E
EU / 1/07/410/049 - 30000IU / 0,75ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 0,75ml (40000IU / ml) 1 seringa pré-cheia de 0,75ml com dispositivo de segurança para Agosto - AIC N. 038190498 / E
EU / 1/07/410/054 - 30.000 UI / 0,75 ML - solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 0,75 ML (40.000 UI / ML) - 4 pré-cheia seringas com dispositivo de segurança para agulha AIC: 038190548 / E
EU / 1/07/410/050 - 30000UI / 0,75ml solução injetável em seringa pré-cheia (vidro) - uso subcutâneo ou intravenoso - 0,75ml (40000UI / ml) 6 seringas pré-cheias de 0,75ml com dispositivo de segurança por agulha - AIC N. 038190500 / E
Binocrit 40.000 UI / 1 mL solução injetável em seringa pré-cheia
EU / 1/07/410/025 - 40.000 UI / 1,0 ml - solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 1,0 ml (40.000 UI / ml) 1 seringa pré-cheia - AIC n. 038190258 / E
EU / 1/07/410/026 - 40.000 UI / 1,0 ml - solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 1,0 ml (40.000 UI / ml) 6 seringas pré-cheias - AIC n. 038190260 / E
EU / 1/07/410/051 - 40000UI / 1ml solução injetável em seringa pré-cheia (vidro) - uso subcutâneo ou intravenoso - 1ml (40000UI / ml) 1 seringa pré-cheia de 1ml com dispositivo de segurança da agulha - AIC N. 038190512 / E
EU / 1/07/410/055 - 40.000 UI / 1.0 ML - solução injetável em seringa pré-cheia - uso subcutâneo ou intravenoso - seringa pré-cheia (vidro) 1.0 ML (40.000 UI / ML) - 4 pré-cheia seringas com agulha de segurança do dispositivo para agulha AIC: 038190551 / E
EU / 1/07/410/052 - 40000UI / 1ml solução injetável em seringa pré-cheia (vidro) - via subcutânea ou intravenosa - 1ml (40000UI / ml) 6 seringas pré-cheias de 1ml com dispositivo de segurança para agulha - AIC N. 038190524 / E
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO -
Desde a primeira autorização: 28 de agosto de 2007
Data da renovação mais recente: 18 de junho de 2012
10.0 DATA DE REVISÃO DO TEXTO -
10/2016