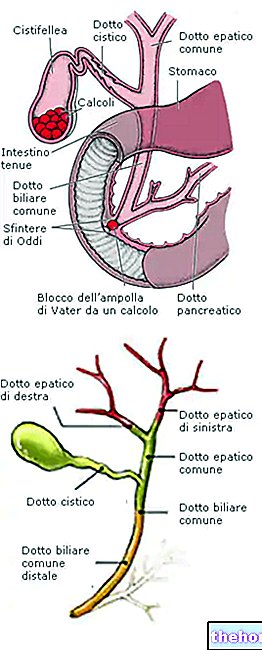
Ingredientes ativos: lenalidomida
Revlimid 2,5 mg cápsulas
Revlimid 5 mg cápsulas
Revlimid cápsulas de 7,5 mg
Revlimid 10 mg cápsulas
Revlimid cápsulas de 15 mg
Revlimid cápsulas de 20 mg
Revlimid 25 mg cápsulas
Por que o Revlimid é usado? Para que serve?
Revlimid contém a substância ativa "lenalidomida". Este medicamento pertence a um grupo de medicamentos que afetam o funcionamento do sistema imunológico.
Revlimid é usado em adultos para:
- Mieloma múltiplo
- Síndromes mielodisplásicas
- Linfoma de células do manto
Mieloma múltiplo e Revlimid
O mieloma múltiplo é um tipo de câncer que afeta um determinado tipo de glóbulos brancos, chamados de células plasmáticas. Essas células se acumulam na medula óssea e se dividem de maneira incontrolável. Isso pode causar danos aos ossos e rins.
O mieloma múltiplo geralmente é incurável. No entanto, os sinais e sintomas podem ser bastante reduzidos ou desaparecer por algum tempo. Este resultado é denominado "resposta".
No tratamento do mieloma múltiplo, Revlimid é usado em combinação com outros medicamentos.
Revlimid em pacientes com mieloma múltiplo recém-diagnosticado
Revlimid é usado apenas em pacientes recém-diagnosticados quando eles não podem ter um transplante de medula óssea.
Se você tem 75 anos de idade ou mais ou tem problemas renais moderados a graves, o seu médico irá avaliá-lo cuidadosamente antes de iniciar o tratamento.
Existem dois tipos de tratamento em pacientes recém-diagnosticados:
- Revlimid juntamente com um medicamento anti-inflamatório denominado “dexametasona”.
- Revlimid juntamente com um medicamento de quimioterapia denominado “melfalano” e um medicamento imunossupressor denominado “prednisona”. Irá tomar estes outros medicamentos no início do tratamento e depois continuará a tomar Revlimid sozinho.
Revlimid em pacientes com mieloma múltiplo que tiveram pelo menos um outro tipo de tratamento antes
- O Revlimid é tomado juntamente com um medicamento anti-inflamatório denominado “dexametasona”.
Revlimid pode impedir o agravamento dos sinais e sintomas de mieloma múltiplo. Também demonstrou atrasar o retorno do mieloma múltiplo após o tratamento.
Síndromes mielodisplásicas e Revlimid
As síndromes mielodisplásicas (SMD) são um conjunto de muitas doenças diferentes do sangue e da medula óssea. As células sanguíneas tornam-se anormais e não funcionam corretamente. Os pacientes podem ter uma variedade de sinais e sintomas, incluindo contagem baixa de glóbulos vermelhos (anemia), necessidade de transfusões de sangue e risco de infecção.
O Revlimid isoladamente é usado para tratar pacientes adultos com diagnóstico de síndromes mielodisplásicas que apresentam todas as seguintes condições:
- se você precisar de transfusões de sangue regulares para tratar níveis baixos de glóbulos vermelhos ("anemia dependente de transfusão")
- se você tem uma "anormalidade nas células da medula óssea chamada" anormalidade citogenética de deleção 5q isolada ". Isso significa que seu corpo não produz células sanguíneas saudáveis suficientes
- se outros tratamentos usados anteriormente são inadequados ou não eficazes o suficiente.
Revlimid pode aumentar o número de glóbulos vermelhos saudáveis produzidos pelo corpo, reduzindo o número de células anormais:
- Isso pode reduzir o número de transfusões de sangue necessárias. Nenhuma transfusão pode ser necessária.
Linfoma de células do manto e Revlimid
O linfoma de células do manto é um câncer do tecido linfático (parte do sistema imunológico), que afeta um tipo de glóbulo branco chamado linfócitos B. No linfoma de células do manto, os linfócitos B crescem descontroladamente e se acumulam no tecido linfático e na medula óssea , ou sangue.
Revlimid é usado isoladamente para tratar pacientes adultos com diagnóstico de linfoma de células do manto não tratado previamente.
Como funciona o Revlimid
Revlimid atua no sistema imunológico do corpo e diretamente no tumor de várias maneiras:
- parando o desenvolvimento de células cancerosas
- parando o crescimento de vasos sanguíneos que transportam sangue para as células tumorais
- estimulando parte do sistema imunológico a atacar as células cancerosas.
Contra-indicações Quando Revlimid não deve ser usado
Não tome Revlimid
- Se está grávida ou pensa estar grávida, ou se planeia engravidar, uma vez que se prevê que Revlimid seja prejudicial para o feto (ver secção 2, “Advertências e precauções” e “Gravidez e amamentação”).
- Se houver possibilidade de engravidar, a menos que tome todas as medidas necessárias para evitar a gravidez (ver secção 2 “Advertências e precauções” e “Gravidez e amamentação”). Se houver a possibilidade de você engravidar, o seu médico observará e confirmará com cada receita que foram tomadas as medidas necessárias para evitar a gravidez.
- Se tem alergia à lenalidomida ou a qualquer outro componente deste medicamento (listados na secção 6). Se você acha que é alérgico, peça conselho ao seu médico.
Se algum destes se aplicar a si, não tome Revlimid. Em caso de dúvida, consulte seu médico.
Precauções de uso O que você precisa saber antes de tomar Revlimid
Informe o seu médico antes de iniciar o tratamento se:
- teve episódios de coágulos sanguíneos no passado, pois o risco de coágulos sanguíneos nas veias e artérias aumenta durante o tratamento
- tem algum sinal de infecção, como tosse ou febre
- tem problemas renais - o seu médico pode alterar a dose de Revlimid
- teve um ataque cardíaco, teve um coágulo sanguíneo ou, se fuma, tem pressão alta ou níveis elevados de colesterol
- tem uma alta carga tumoral em todo o corpo, incluindo na medula óssea. Isso pode levar a uma doença em que os tumores se rompem e causam níveis incomuns de substâncias químicas no sangue, o que pode levar à insuficiência renal (esta doença é chamada de "tumor síndrome de lise ")
- tiveram uma reação alérgica durante o tratamento com talidomida, como erupção na pele, coceira, inchaço, tontura ou dificuldade em respirar
Se algum destes se aplicar a si, informe o seu médico antes de iniciar o tratamento.
Se você tem síndromes mielodisplásicas, é mais provável que desenvolva uma doença mais avançada chamada leucemia mieloide aguda (LMA). Além disso, o papel do Revlimid na probabilidade de desenvolver LMA não é conhecido.O seu médico pode pedir-lhe alguns testes para verificar se há sinais que possam prever com mais precisão a probabilidade de desenvolver LMA durante o tratamento com Revlimid.
Análise e controles
Irá fazer análises sanguíneas regulares antes e durante o tratamento com Revlimid, uma vez que Revlimid pode causar uma redução nas células sanguíneas que o defendem contra infecções (glóbulos brancos) e aquelas que ajudam o sangue a coagular (plaquetas). O seu médico irá pedir-lhe para fazer uma análise ao sangue:
- antes do tratamento
- todas as semanas durante as primeiras 8 semanas de tratamento (para pacientes com linfoma de células do manto, isso acontecerá a cada 2 semanas nos ciclos 3 e 4 e, em seguida, no início de cada ciclo)
- depois disso, pelo menos uma vez por mês.
Seu médico pode verificar se há alterações em sua pele, como manchas vermelhas ou erupções cutâneas.
O seu médico pode decidir ajustar a dose de Revlimid ou interromper o tratamento, dependendo dos resultados das análises ao sangue e do seu estado geral. Se você for um paciente recém-diagnosticado, seu médico também pode avaliar o tratamento com base na sua idade e em quaisquer outras condições que já possam estar presentes.
Doação de sangue
Você não deve doar sangue durante a terapia com lenalidomida e por uma semana após interromper o tratamento.
Crianças e adolescentes
Revlimid não é recomendado para uso em crianças e adolescentes com menos de 18 anos de idade.
Interações Quais medicamentos ou alimentos podem alterar o efeito do Revlimid
Informe o seu médico ou enfermeiro se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos, incluindo medicamentos obtidos sem receita médica e medicamentos à base de plantas, porque Revlimid pode afetar o modo como alguns outros medicamentos atuam e alguns outros medicamentos podem afetar o forma como o Revlimid funciona.
Em particular, informe o seu médico ou enfermeiro se estiver tomando algum dos seguintes medicamentos:
- alguns medicamentos usados para prevenir a gravidez, como anticoncepcionais orais, pois podem não ser mais eficazes
- alguns medicamentos usados para problemas cardíacos, como a digoxina
- alguns medicamentos usados para tornar o sangue mais fluido, como a varfarina
Avisos É importante saber que:
Gravidez, amamentação e contracepção - informações para mulheres e homens
Gravidez
Para mulheres que tomam Revlimid
- Não deve tomar Revlimid se estiver grávida, uma vez que se prevê que este medicamento seja prejudicial para o feto.
- Não deve engravidar durante o tratamento com Revlimid. Se houver possibilidade de gravidez, você deve usar métodos eficazes de contracepção (ver seção “Contracepção”).
- Se engravidar durante o tratamento com Revlimid, deve interromper o tratamento imediatamente e informar o seu médico.
Para homens tomando Revlimid
- Se a sua parceira engravidar enquanto estiver a tomar Revlimid, informe o seu médico imediatamente. Também é recomendado que seu parceiro entre em contato com seu médico.
- Além disso, você deve usar métodos eficazes de contracepção (consulte a seção “Contracepção”).
Hora da alimentação
Não deve amamentar enquanto está a tomar Revlimid, uma vez que não se sabe se este medicamento passa para o leite materno.
Contracepção
Para mulheres que tomam Revlimid
Antes de iniciar o tratamento, deve perguntar ao seu médico se existe a possibilidade de engravidar, mesmo que pense ser improvável.
Se houver a possibilidade de você ficar grávida
- você terá que se submeter a testes de gravidez sob a supervisão do seu médico (antes de cada tratamento, a cada 4 semanas durante o tratamento e 4 semanas após o final do tratamento), exceto nos casos em que foi confirmado que as trompas de falópio foram cortadas e fechadas, para prevenir os ovos de atingir o útero (esterilização por amarrar as trompas)
- deve usar métodos contraceptivos eficazes durante 4 semanas antes do início do tratamento, durante o tratamento e até 4 semanas após a interrupção do tratamento.O seu médico irá aconselhá-la sobre os métodos contraceptivos adequados.
Para homens tomando Revlimid
Revlimid passa para o sêmen humano. Se houver a possibilidade de sua parceira estar grávida ou engravidar e ela não estiver usando métodos anticoncepcionais eficazes, você deve usar preservativo durante o tratamento e por uma semana após o término do tratamento, mesmo que você tenha feito uma vasectomia.
Condução e utilização de máquinas
Não conduza nem opere máquinas se sentir tonturas, cansaço, sonolência, tonturas ou visão turva.
Revlimid contém lactose
Revlimid contém lactose. Se foi informado pelo seu médico que tem "intolerância a alguns açúcares, contacte-o antes de tomar Revlimid.
Dose, método e tempo de administração Como usar Revlimid: Posologia
Revlimid deve ser administrado por profissionais de saúde com experiência no tratamento de mieloma múltiplo ou síndromes mielodisplásicas e linfoma de células do manto.
- Quando utilizado para tratar mieloma múltiplo, Revlimid é tomado em associação com outros medicamentos (ver secção 1 “O que é“ Revlimid e para que é utilizado ”).
- Quando utilizado para o tratamento de síndromes mielodisplásicas e linfoma de células do manto, o Revlimid é tomado isoladamente.
Sempre tome Revlimid sozinho ou Revlimid em combinação com outros medicamentos, sempre seguindo exatamente as instruções do médico. Em caso de dúvida, consulte o seu médico ou farmacêutico.
Se estiver a tomar Revlimid em associação com outros medicamentos, consulte os folhetos informativos desses medicamentos para mais informações sobre a sua utilização e efeitos.
Ciclo de tratamento
- Revlimid e os medicamentos que precisa de tomar em combinação com Revlimid são tomados ao longo de alguns dias ao longo de um período de 4 semanas (28 dias).
- Cada período de 28 dias é denominado "ciclo de tratamento".
- Dependendo do dia da sua menstruação, você tomará um ou mais dos medicamentos. No entanto, em alguns dias você não tomará nenhum medicamento.
- Assim que cada ciclo de 28 dias for concluído, um novo "ciclo" deve começar nos próximos 28 dias.
Dose de Revlimid a tomar
Antes de iniciar o tratamento, o seu médico irá dizer-lhe:
- a dose de Revlimid a tomar
- a dose de outros medicamentos a serem tomados em combinação com Revlimid, se prescrito
- em que dias do ciclo de tratamento deve tomar cada medicamento.
O médico pode observar alterações na pele, como manchas vermelhas ou erupções cutâneas.
O seu médico também pode decidir alterar a dose de Revlimid ou de outros medicamentos durante o tratamento, com base nos resultados das análises ao sangue e no seu estado geral (ver secção 2, “O que precisa de saber antes de tomar Revlimid”).
Como e quando tomar Revlimid
- As cápsulas devem ser engolidas inteiras, de preferência com água.
- Não quebre, abra ou mastigue as cápsulas.
- As cápsulas podem ser tomadas com ou sem alimentos.
- Deve tomar Revlimid nos dias marcados aproximadamente à mesma hora.
Para retirar a cápsula do blister, pressione apenas um dos lados da cápsula, empurrando-a através da folha de alumínio. Não pressione o centro da cápsula, caso contrário pode partir-se.
Duração do tratamento com Revlimid
O Revlimid é administrado em ciclos de tratamento, cada um com a duração de 28 dias (ver “Ciclo de tratamento” acima). Deve continuar o tratamento até que o seu médico lhe diga para parar o tratamento.
Caso se tenha esquecido de tomar Revlimid
Se você se esquecer de tomar Revlimid no horário normal e
- menos de 12 horas se passaram: tome sua cápsula imediatamente
- passaram mais de 12 horas: não tome a cápsula esquecida, mas tome a cápsula seguinte no dia seguinte à hora habitual.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
Sobredosagem O que fazer se você tiver tomado muito Revlimid
Se tomou mais Revlimid do que lhe foi receitado, informe o seu médico imediatamente.
Efeitos colaterais Quais são os efeitos colaterais do Revlimid
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
Efeitos colaterais graves que podem afetar mais de 1 em 10 pessoas
Revlimid pode reduzir o número de glóbulos brancos que combatem infecções e células sanguíneas que promovem a coagulação do sangue (plaquetas), o que pode causar distúrbios hemorrágicos, por ex. sangramento nasal e hematomas. Revlimid também pode causar coágulos sanguíneos nas veias (trombose).
Portanto, você deve procurar atendimento médico imediato se sentir algum dos seguintes efeitos colaterais:
- febre, calafrios, dor de garganta, tosse, úlceras na boca ou quaisquer outros sintomas de infecção (incluindo na corrente sanguínea (sepse))
- sangramento ou hematomas na ausência de feridas
- dor no peito ou nas pernas
- falta de ar.
Se sentir algum dos efeitos secundários listados acima, informe o seu médico imediatamente.
Outros efeitos colaterais estão listados abaixo
É importante notar que um pequeno número de pacientes pode desenvolver outros tipos de câncer e é possível que esse risco aumente com o tratamento com Revlimid; portanto, o seu médico deve pesar cuidadosamente os benefícios e riscos ao prescrever Revlimid para você.
Efeitos colaterais muito comuns podem afetar mais de 1 em 10 pessoas:
- Diminuição do número de glóbulos vermelhos (anemia), que pode causar cansaço e fraqueza
- Constipação, diarreia, náusea, vermelhidão da pele, erupção cutânea, vômito, cãibras musculares, dores musculares, dor óssea, dor nas articulações, fadiga, inchaço generalizado, incluindo inchaço dos braços e pernas
- Febre e sintomas de gripe, incluindo febre, dor muscular, dor de cabeça, dor de ouvido e calafrios
- Dormência, formigamento ou sensação de queimação na pele, dor nas mãos ou pés, tontura, tremor, alteração no paladar
- Dor no peito com irradiação para os braços, pescoço, mandíbula, costas ou estômago, com sensação de suor e falta de ar, náuseas ou vômitos, que podem ser sintomas de um ataque cardíaco (infarto do miocárdio)
- Redução do apetite
- Níveis baixos de potássio no sangue
- Dor nas pernas (que pode ser um sintoma de trombose), dor no peito ou falta de ar (que podem ser sintomas de coágulos sanguíneos nos pulmões, chamados de embolia pulmonar)
- Infecções de qualquer tipo
- Infecção dos pulmões e do trato respiratório superior, falta de ar
- Visão embaçada
- Visão turva (catarata)
- Problemas renais
- Alterações em uma proteína do sangue que podem causar inchaço das artérias (vasculite)
- Aumentos de açúcar no sangue (diabetes)
- Dor de cabeça
- Pele seca
- Dor de estômago
- Mudança de humor, dificuldade para dormir
Os efeitos colaterais comuns podem afetar até 1 em cada 10 pessoas:
- Infecção dos seios da face ao redor do nariz
- Sangramento da gengiva, estômago ou intestinos
- Aumento da dor, tamanho do tumor, vermelhidão ao redor do tumor
- Aumento ou diminuição da pressão arterial, batimento cardíaco lento, rápido ou irregular
- Escurecimento da pele
- Erupções cutâneas, pele rachada, descamação ou descamação
- Urticária, coceira, aumento da sudorese, desidratação
- Boca inflamada com úlceras, boca seca, dificuldade em engolir
- Dor de estômago
- Produção de urina muito mais ou menos do que o normal (o que pode ser um sintoma de insuficiência renal), sangue na urina
- Falta de ar, especialmente quando deitado (que pode ser um sintoma de insuficiência cardíaca)
- Dificuldade em obter uma ereção
- AVC, desmaio
- Fraqueza muscular
- Inchaço das articulações
- Alterações no hormônio da tireoide no sangue, níveis baixos de cálcio, fosfato ou magnésio no sangue
- Depressão
- Surdez
- Testes de função hepática anormais
- Distúrbios de equilíbrio, dificuldades de movimento
- Zumbido nos ouvidos (zumbido)
- Sobrecarga de ferro
- Sede
- Confusão
- Dor de dente
- Perda de peso.
Os efeitos colaterais incomuns podem afetar até 1 em 100 pessoas:
- Hemorragia dentro do crânio
- Problemas circulatórios
- Perda de visão
- Perda do desejo sexual (libido)
- Fluxo abundante de urina com dor e fraqueza nos ossos, que podem ser sintomas de uma doença renal (síndrome de Fanconi)
- Dor de estômago, distensão abdominal ou diarreia, que podem ser sintomas de inflamação do intestino grosso (chamada colite ou tiflite)
- Produção de muito mais ou menos urina do que o normal, o que pode ser um sintoma de um tipo de problema renal (denominado necrose tubular renal
- Descoloração da pele, sensibilidade à luz solar
- Alguns tipos de câncer de pele
- Urticária, erupção na pele, inchaço dos olhos, boca ou face, dificuldade em respirar ou coceira, que podem ser sintomas de uma reação alérgica.
Efeitos colaterais raros podem afetar até 1 em 1.000 pessoas:
- Reação alérgica grave, que pode começar como erupção cutânea em uma área, mas se espalhar com extensa perda de pele por todo o corpo (síndrome de Stevens-Johnson e / ou necrólise epidérmica tóxica).
- Síndrome de lise tumoral - complicações metabólicas que podem ocorrer durante o tratamento do tumor e às vezes até mesmo sem tratamento. Essas complicações são causadas pelos produtos da degradação das células cancerosas que estão morrendo e podem incluir as seguintes complicações: alterações nos parâmetros hematológicos; altos valores de potássio, fósforo e ácido úrico; e baixos valores de cálcio que, consequentemente, conduzem a alterações da função renal, frequência cardíaca, convulsões e, por vezes, morte.
Frequência desconhecida: a frequência não pode ser estimada a partir dos dados disponíveis:
- Dor súbita ou leve, mas com piora na parte superior do abdômen e / ou nas costas, que persiste por alguns dias, possivelmente com náuseas, vômitos, febre e pulso rápido. Esses sintomas podem ser causados por inflamação do pâncreas.
- Chiado, falta de ar ou tosse seca, que podem ser sintomas causados por inflamação do tecido pulmonar.
- Descoloração amarelada da pele, membranas mucosas ou olhos (icterícia), fezes de cor clara, urina de cor escura, coceira na pele, erupção cutânea, dor ou inchaço do abdômen. Estes podem ser sintomas de lesão hepática (doença hepática).
- Foram observados casos raros de destruição muscular (dor, fraqueza ou inchaço dos músculos), que podem causar problemas renais (rabdomiólise), alguns dos quais quando Revlimid é administrado com uma estatina (um tipo de medicamento para baixar o colesterol).
- Doença que afeta a pele e é causada pela inflamação dos pequenos vasos sanguíneos, com dor nas articulações e febre (vasculite leucocitoclástica).
- Deterioração do estômago ou da parede intestinal, que pode causar infecções muito graves.Informe o seu médico se você tiver fortes dores abdominais, febre, náuseas, vômitos, sangue nas fezes ou alterações nos hábitos intestinais.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico, farmacêutico ou enfermeiro. Isto inclui quaisquer efeitos secundários possíveis não listados neste folheto. Também pode comunicar os efeitos secundários diretamente através do sistema nacional de notificação listado no Apêndice V. efeitos secundários pode ajudar fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
- Mantenha este medicamento fora da vista e do alcance das crianças.
- Não utilize este medicamento após o prazo de validade impresso no blister e na embalagem exterior após “VAL”. A data de validade refere-se ao último dia desse mês.
- Este medicamento não requer quaisquer condições especiais de armazenamento.
- Não use este medicamento se notar que as embalagens estão danificadas ou mostram sinais de adulteração.
- Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Prazo "> Outras informações
O que Revlimid contém
Revlimid cápsulas de 2,5 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 2,5 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina, dióxido de titânio (E171), índigo carmim (E132) e óxido de ferro amarelo (E172)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid 5 mg cápsulas:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 5 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina e dióxido de titânio (E171)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid cápsulas de 7,5 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 7,5 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina, dióxido de titânio (E171), óxido de ferro amarelo (E172)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid cápsulas de 10 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 10 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- preparação da cápsula: gelatina, dióxido de titânio (E171), índigo carmim (E132) e óxido de ferro amarelo (E172)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid cápsulas de 15 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 15 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina, dióxido de titânio (E171) e índigo carmim (E132)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid cápsulas de 20 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 20 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina, dióxido de titânio (E171), índigo carmim (E132) e óxido de ferro amarelo (E172)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Revlimid cápsulas de 25 mg:
- O ingrediente ativo é lenalidomida. Cada cápsula contém 25 mg de lenalidomida.
- Os outros ingredientes são:
- conteúdo da cápsula: lactose anidra, celulose microcristalina, croscarmelose sódica e estearato de magnésio
- invólucro da cápsula: gelatina e dióxido de titânio (E171)
- tinta para letras: goma laca, propilenoglicol, hidróxido de potássio e óxido de ferro preto (E172).
Qual a aparência de Revlimid e conteúdo da embalagem
As cápsulas de Revlimid 2,5 mg são azul-esverdeadas / brancas, marcadas com “REV 2,5 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo um ou três blisters. Cada blister contém sete cápsulas, para um total de 7 ou 21 cápsulas por embalagem.
As cápsulas de Revlimid 5 mg são brancas, marcadas “REV 5 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo um ou três blisters. Cada blister contém sete cápsulas, para um total de 7 ou 21 cápsulas por embalagem.
As cápsulas de Revlimid de 7,5 mg são amarelo pálido / branco, marcadas com “REV 7,5 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo três blisters. Cada blister contém sete cápsulas, para um total de 21 cápsulas por embalagem.
As cápsulas de Revlimid de 10 mg são azul-esverdeadas / amarelo claro, marcadas com “REV 10 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo três blisters. Cada blister contém sete cápsulas, para um total de 21 cápsulas por embalagem.
As cápsulas de Revlimid 15 mg são azuis claras / brancas, marcadas com “REV 15 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo três blisters. Cada blister contém sete cápsulas, para um total de 21 cápsulas por embalagem.
As cápsulas de Revlimid 20 mg são azul esverdeado / azul claro, marcadas com “REV 20 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo três blisters. Cada blister contém sete cápsulas, para um total de 21 cápsulas por embalagem.
As cápsulas de Revlimid 25 mg são brancas, marcadas com “REV 25 mg”.
As cápsulas são fornecidas em embalagens, cada uma contendo três blisters. Cada blister contém sete cápsulas, para um total de 21 cápsulas por embalagem.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO -
CÁPSULAS DURAS REVLIMID 10 MG
▼ Medicamento sujeito a monitorização adicional. Isso permitirá a rápida identificação de novas informações de segurança. Os profissionais de saúde são solicitados a notificar quaisquer suspeitas de reações adversas. Consulte a seção 4.8 para obter informações sobre como notificar reações adversas.
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA -
Cada cápsula contém 10 mg de lenalidomida.
Excipientes com efeitos conhecidos:
Cada cápsula contém 294 mg de lactose anidra.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA -
Cápsula dura.
Cápsulas azul esverdeado / amarelo claro, tamanho 0,21,7 mm, marcadas "REV 10 mg".
04.0 INFORMAÇÕES CLÍNICAS -
04.1 Indicações terapêuticas -
Mieloma múltiplo
Revlimid está indicado para o tratamento de doentes adultos com mieloma múltiplo não tratado anteriormente e que não são elegíveis para transplante (ver secção 4.2).
Revlimid, em combinação com dexametasona, é indicado para o tratamento de pacientes adultos com mieloma múltiplo que receberam pelo menos uma terapia anterior.
Síndromes mielodisplásicas
Revlimid é indicado para o tratamento de pacientes com anemia dependente de transfusão devido a síndromes mielodisplásicas (SMD) de risco baixo ou intermediário-1 associadas a anormalidade citogenética de deleção 5q isolada, quando outras opções de tratamento são insuficientes ou inadequadas.
Linfoma de células do manto
Revlimid está indicado para o tratamento de doentes adultos com linfoma de células do manto recidivante ou refractário (ver secções 4.4 e 5.1).
04.2 Posologia e método de administração -
O tratamento com Revlimid deve ser supervisionado por um médico com experiência na utilização de terapias contra o cancro (ver secção 4.4, cariótipo).
Dosagem
Mieloma múltiplo recentemente diagnosticado
Lenalidomida em combinação com dexametasona até a progressão da doença, em não pacientes elegível para transplante
O tratamento com lenalidomida não deve ser iniciado se a contagem absoluta de neutrófilos (Absoluto
Contagem de neutrófilos, ANC) é
Dose recomendada
A dose inicial recomendada de lenalidomida é de 25 mg por via oral uma vez ao dia nos dias 1-21 dos ciclos repetidos de 28 dias. A dose recomendada de dexametasona é de 40 mg por via oral uma vez ao dia nos dias 1, 8, 15 e 22 de ciclos repetidos de 28 dias. Os pacientes podem continuar a terapia com lenalidomida e dexametasona até o desenvolvimento de progressão da doença ou intolerância.
A posologia pode ser continuada ou modificada com base nas conclusões clínicas e laboratoriais (ver secção 4.4). Para pacientes ≥ 75 anos de idade, a dose inicial de dexametasona é de 20 mg / dia nos dias 1, 8, 15 e 22 de cada ciclo de tratamento de 28 dias. A dose recomendada de lenalidomida para pacientes com insuficiência renal moderada é de 10 mg uma vez ao dia.
Ajustes de dose recomendados durante o tratamento e após o reinício do tratamento
Conforme resumido nas tabelas abaixo, os ajustes de dose são recomendados para o tratamento da trombocitopenia e neutropenia de Grau 3 ou 4, ou para o tratamento de quaisquer outras toxicidades de Grau 3 ou 4 que se acredita estarem relacionadas com a lenalidomida.
• Níveis de redução de dose
• Trombocitopenia
a Se ocorrer toxicidade limitante da dose (Toxicidade Limitadora de Dose, DLT)> Dia 15 de um ciclo, a dosagem de lenalidomida será interrompida pelo menos pelo restante do ciclo atual de 28 dias.
• Neutropenia
Em caso de neutropenia, o médico deve considerar o uso de fatores de crescimento no tratamento do paciente.
Se a dose de lenalidomida foi reduzida devido ao DLT hematológico, a dose de lenalidomida pode ser reintroduzida no próximo nível de dose mais alto (até a dose inicial), a critério do médico assistente, se a terapia continuada com lenalidomida / dexametasona produziu melhora na medula óssea função (ausência de DLT por pelo menos 2 ciclos consecutivos e ANC ≥ 1.500 / mcl, com contagem de plaquetas ≥ 100.000 / mcl, no início de um novo ciclo na dose atual).
Lenalidomida em combinação com melfalano e prednisona, seguida por monoterapia de manutenção, em pacientes não elegíveis para transplante
O tratamento com lenalidomida não deve ser iniciado se o ANC for
Dose recomendada
A dose inicial recomendada é lenalidomida 10 mg / dia por via oral nos dias 1-21 de ciclos repetidos de 28 dias por até 9 ciclos, melfalan 0,18 mg / kg por via oral nos dias 1-4 de ciclos repetidos de 28 dias, prednisona 2 mg / kg por via oral nos dias 1-4 de ciclos repetidos de 28 dias. Os pacientes que completam 9 ciclos ou que não conseguem completar a terapia combinada devido à intolerância devem ser tratados com lenalidomida em monoterapia, 10 mg / dia por via oral, nos dias 1-21 de ciclos repetidos 28 dias até a progressão da doença. A posologia pode ser continuada ou modificada com base nas conclusões clínicas e laboratoriais (ver secção 4.4).
Ajustes de dose recomendados durante o tratamento e após o reinício do tratamento
Conforme resumido nas tabelas abaixo, os ajustes de dose são recomendados para o tratamento de trombocitopenia de Grau 3 ou 4 ou neutropenia, ou para o tratamento de qualquer outra toxicidade de Grau 3 ou 4 que se pensa estar relacionada à lenalidomida.
• Níveis de redução de dose
Se a neutropenia for a única toxicidade em qualquer dosagem, adicionar fator estimulador de colônia de granulócitos (G-CSF) e manter a dosagem de lenalidomida..
• Trombocitopenia
• Neutropenia
a Se o indivíduo não recebeu terapia com G-CSF, inicie a terapia com G-CSF. No dia 1 do próximo ciclo, continue GCSF conforme necessário e mantenha a dose de melfalan se a neutropenia foi o único DLT. Caso contrário, reduza um nível de dose no início do próximo ciclo.
Em caso de neutropenia, o uso de fatores de crescimento no tratamento do paciente deve ser considerado.
Mieloma múltiplo com pelo menos uma terapia anterior
Dose recomendada
A dose inicial recomendada é de 25 mg de lenalidomida por via oral uma vez ao dia nos dias 1-21 dos ciclos repetidos de 28 dias. A dose recomendada de dexametasona é de 40 mg por via oral uma vez ao dia nos dias 1-4, 9-12 e 17-20 de cada ciclo de 28 dias para os primeiros 4 ciclos de terapia e 40 mg uma vez ao dia a partir de então. a cada 28 dias.
A posologia pode ser continuada ou modificada com base nas conclusões clínicas e laboratoriais (ver secção 4.4). Os médicos devem avaliar cuidadosamente a dosagem de dexametasona a ser usada, levando em consideração a condição do paciente e o estado da doença.
O tratamento com lenalidomida não deve ser iniciado se o ANC for medula óssea por células plasmáticas, se a contagem de plaquetas for
Ajustes de dose recomendados durante o tratamento e após o reinício do tratamento
Conforme resumido nas tabelas abaixo, os ajustes de dose são recomendados para o tratamento de neutropenia ou trombocitopenia de grau 3 ou 4, ou para o tratamento de qualquer toxicidade de grau 3 ou 4 que se pensa estar relacionada à lenalidomida.
• Níveis de redução de dose
• Trombocitopenia
• Neutropenia
Em caso de neutropenia, o uso de fatores de crescimento no tratamento do paciente deve ser considerado.
Síndromes mielodisplásicas
O tratamento com lenalidomida não deve ser iniciado se o ANC for
Dose recomendada
A dose inicial recomendada é de 10 mg de lenalidomida por via oral uma vez ao dia nos dias 1-21 dos ciclos repetidos de 28 dias. A posologia pode ser continuada ou modificada com base nas conclusões clínicas e laboratoriais (ver secção 4.4).
Ajustes de dose recomendados durante o tratamento e após o reinício do tratamento
Conforme resumido nas tabelas abaixo, os ajustes de dose são recomendados para o tratamento de neutropenia ou trombocitopenia de grau 3 ou 4, ou para o tratamento de qualquer toxicidade de grau 3 ou 4 que se pensa estar relacionada à lenalidomida.
• Níveis de redução de dose
Para pacientes começando com uma dose de 10 mg e apresentando trombocitopenia ou neutropenia:
• Trombocitopenia
• Neutropenia
Suspensão de lenalidomida
Pacientes que não apresentam pelo menos uma resposta eritróide leve dentro de 4 meses do início da terapia, demonstrada por uma redução de pelo menos 50% nas necessidades de transfusão ou, se não estiverem recebendo transfusões, por um aumento de 1 g / dl na hemoglobina, devem interromper a lenalidomida tratamento.
Linfoma de células do manto
Dose recomendada
A dose inicial recomendada é de 25 mg de lenalidomida por via oral uma vez ao dia nos dias 1-21 dos ciclos repetidos de 28 dias.
O ensaio é continuado ou modificado com base nos resultados clínicos e laboratoriais (ver secção 4.4).
Ajustes de dose recomendados durante o tratamento e após o reinício do tratamento
Conforme resumido nas tabelas abaixo, os ajustes de dose são recomendados para o tratamento de neutropenia ou trombocitopenia de Grau 3 ou 4, ou para o tratamento de qualquer toxicidade de Grau 3 ou 4 que se pensa estar relacionada à lenalidomida.
• Níveis de redução de dose
1 - Em países onde a cápsula de 2,5 mg está disponível.
• Trombocitopenia
• Neutropenia
• Reação de exacerbação do tumor
O tratamento com lenalidomida pode ser continuado em pacientes com Reação de alargamento de tumor, Indenização por desligamento de grau 1 ou 2, sem interrupção ou modificação, a critério do médico. Em pacientes com TFR de Grau 3 ou 4, o tratamento com lenalidomida deve ser suspenso até que a TFR seja reduzida para ≤ Grau 1; Para a gestão dos sintomas, os doentes podem ser tratados de acordo com as directrizes de TFR de grau 1 e 2 (ver secção 4.4).
Todos os pacientes
Para outros efeitos tóxicos de Grau 3 ou 4 que se acredita estarem relacionados com a lenalidomida, o tratamento deve ser descontinuado e retomado na próxima dose mais baixa apenas quando a toxicidade tiver diminuído para ≤ Grau 2 a critério do médico.
A descontinuação ou descontinuação da lenalidomida deve ser considerada em caso de erupção cutânea de grau 2 ou 3. O tratamento com lenalidomida deve ser interrompido em caso de angioedema, erupção cutânea de grau 4, erupção cutânea esfoliativa ou bolhosa, ou se houver suspeita de Stevens-Johnson (SSJ) ou epidérmico tóxico necrólise (NET), e não deve ser retomado após a interrupção devido a essas reações.
Populações especiais
População pediátrica
Revlimid não deve ser utilizado em crianças e adolescentes desde o nascimento até aos 18 anos de idade devido a questões de segurança (ver secção 4.4).
Pacientes idosos
Os dados farmacocinéticos atualmente disponíveis são descritos na seção 5.2. A lenalidomida foi utilizada em estudos clínicos em doentes com mieloma múltiplo até 91 anos de idade, em doentes com síndromes mielodisplásicas até 95 anos e em doentes com linfoma de células do manto até 88 anos de idade (ver secção 5.1).
Em pacientes com mieloma múltiplo recém-diagnosticados com 75 anos de idade ou mais tratados com lenalidomida, houve uma maior incidência de reações adversas graves e reações adversas que levaram à descontinuação do tratamento (ver seção 4.4). Pacientes com mieloma múltiplo recém-diagnosticados com 75 anos de idade ou mais devem ser cuidadosamente avaliado antes de considerar o tratamento (ver secção 4.4).
• Mieloma múltiplo recém-diagnosticado
Para pacientes com mais de 75 anos de idade tratados com lenalidomida em combinação com dexametasona, a dose inicial de dexametasona é de 20 mg / dia nos dias 1, 8, 15 e 22 de cada ciclo de tratamento de 28 dias.
Não são propostos ajustes de dose para pacientes com mais de 75 anos de idade tratados com lenalidomida em combinação com melfalano e prednisona.
Em ensaios clínicos de mieloma múltiplo recentemente diagnosticado em doentes não elegíveis para transplante, a terapêutica combinada com lenalidomida foi menos tolerada em doentes com mais de 75 anos de idade do que na população mais jovem. Entre esses pacientes, a porcentagem que interrompeu o tratamento devido à intolerância (eventos adversos de grau 3 ou 4 e eventos adversos graves) foi maior do que em pacientes com idade
• Mieloma múltiplo tratado anteriormente com pelo menos uma terapia
A porcentagem de pacientes com mieloma múltiplo com 65 anos ou mais não foi significativamente diferente entre os grupos lenalidomida / dexametasona e placebo / dexametasona. Em geral, não foram observadas diferenças de segurança e eficácia entre esses pacientes e os pacientes mais jovens, embora uma maior predisposição de pacientes mais velhos não possa ser descartada.
• Síndromes mielodisplásicas
Para pacientes com síndromes mielodisplásicas tratados com lenalidomida, nenhuma diferença geral na segurança e eficácia foi observada entre pacientes com mais de 65 anos de idade e pacientes mais jovens.
• Linfoma de células do manto
Para pacientes com linfoma de células do manto tratados com lenalidomida, nenhuma diferença geral na segurança e eficácia foi observada entre pacientes com 65 anos ou mais e pacientes com menos de 65 anos.
Como os pacientes idosos são mais propensos a ter função renal comprometida, cuidado especial deve ser tomado na seleção da dose e a monitoração da função renal deve ser realizada como precaução.
Pacientes com insuficiência renal
A lenalidomida é substancialmente excretada pelos rins; em doentes com graus mais elevados de insuficiência renal, a tolerabilidade do tratamento pode ser alterada (ver secção 4.4). Deve-se ter um cuidado especial na escolha da posologia e é aconselhável monitorar a função renal.
Não é necessário ajuste de dose em pacientes com insuficiência renal leve e mieloma múltiplo, síndromes mielodisplásicas ou linfoma de células do manto. Em pacientes com insuficiência renal moderada ou grave ou doença renal em estágio final, no início da terapia e ao longo da duração do tratamento, os seguintes ajustes de dose são recomendados: Não há experiência de ensaio clínico de fase III em pacientes com doença renal em estágio final (ESRD ) (Diálise CLcr).
• Mieloma múltiplo
1 A dose pode ser aumentada para 15 mg uma vez ao dia após 2 ciclos se o paciente não responder ao tratamento, mas tolerar o medicamento.
2 Em países onde a cápsula de 7,5 mg está disponível.
• Síndromes mielodisplásicas
* Níveis de redução de dose recomendados durante o tratamento e após o reinício do tratamento, para controlar a neutropenia ou trombocitopenia de grau 3 ou 4 ou outra toxicidade de grau 3 ou 4 considerada relacionada à lenalidomida, conforme descrito acima.
• Linfoma de células do manto
1 A dose pode ser aumentada para 15 mg uma vez ao dia após 2 ciclos se o paciente não responder ao tratamento, mas tolerar o medicamento.
2 Em países onde a cápsula de 7,5 mg está disponível.
Após o início da terapia com lenalidomida, a modificação subsequente da dose de lenalidomida em pacientes com insuficiência renal deve ser baseada na tolerabilidade do tratamento para o paciente individual, conforme descrito acima.
Pacientes com insuficiência hepática
A lenalidomida não foi estudada formalmente em pacientes com insuficiência hepática e não há recomendações posológicas específicas.
Método de administração
Uso oral.
As cápsulas de Revlimid devem ser tomadas nos dias designados, aproximadamente à mesma hora. As cápsulas não devem ser abertas, partidas ou mastigadas. As cápsulas devem ser engolidas inteiras, de preferência com água, com ou sem alimentos. O paciente pode tomar uma dose esquecida se for menos de 12 horas após o horário programado para tomá-la. Se, por outro lado, for mais de 12 horas, o paciente não deve tomar a dose esquecida, mas esperar o usual hora do dia seguinte para tomar a próxima dose.
Recomenda-se aplicar pressão em apenas um dos lados da cápsula para retirá-la do blister, reduzindo o risco de deformá-la ou quebrá-la.
04.3 Contra-indicações -
• Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na secção 6.1.
• Mulheres grávidas.
• Mulheres com potencial para engravidar, a menos que sejam cumpridas todas as condições do Programa de Prevenção da Gravidez (ver secções 4.4 e 4.6).
04.4 Advertências especiais e precauções adequadas de uso -
Precauções em caso de gravidez
A lenalidomida está estruturalmente relacionada com a talidomida, uma substância ativa com conhecido efeito teratogênico em humanos, que causa malformações congênitas graves com risco de vida. Malformações induzidas pela lenalidomida em macacos semelhantes às descritas para a talidomida (ver seções 4.6 e 5.3). Um efeito teratogênico da lenalidomida é esperado em humanos durante a gravidez.
As condições do Programa de Prevenção de Gravidez devem ser atendidas para todas as pacientes, a menos que haja evidências firmes de que a paciente é incapaz de conceber.
Critérios para estabelecer que uma mulher não tem potencial para engravidar
Uma paciente do sexo feminino ou o parceiro de um paciente do sexo masculino é considerado capaz de engravidar, a menos que ela atenda a pelo menos um dos seguintes critérios:
• Idade ≥ 50 anos e amenorreia natural * por ≥ 1 ano
• Insuficiência ovariana prematura confirmada por um ginecologista
• Salpingo-ooforectomia ou histerectomia bilateral anterior
• Genótipo XY, síndrome de Turner, agenesia uterina.
* Amenorréia após terapia anticâncer ou durante a amamentação não exclui a fertilidade potencial.
Orientação
Lenalidomida é contra-indicada para mulheres com potencial para engravidar, a menos que todas as seguintes condições sejam atendidas:
• O paciente está ciente de que é esperado um risco teratogênico para o feto
• A paciente está ciente da necessidade de usar métodos anticoncepcionais eficazes, sem interrupção, 4 semanas antes do início do tratamento, durante toda a duração do tratamento e até 4 semanas após o término do tratamento.
• Mesmo na presença de amenorreia, uma paciente com potencial para engravidar deve seguir todas as recomendações de contracepção eficaz
• O paciente deve ser capaz de cumprir medidas anticoncepcionais eficazes
• A paciente é informada e está ciente das possíveis consequências da gravidez e da necessidade de procurar atendimento médico imediato se houver risco de gravidez
• A paciente está ciente da necessidade de iniciar o tratamento assim que a lenalidomida for dispensada após um teste de gravidez negativo
• A paciente está ciente da necessidade e concorda em realizar testes de gravidez a cada 4 semanas, exceto nos casos de esterilização confirmada por laqueadura
• A paciente reconhece que está ciente dos riscos e precauções necessárias associadas ao uso de lenalidomida
Para pacientes do sexo masculino que tomam lenalidomida, os estudos farmacocinéticos demonstraram que, durante o tratamento, a lenalidomida está presente em níveis extremamente baixos no sêmen e é indetectável no sêmen de indivíduos saudáveis 3 dias após a interrupção da substância (ver parágrafo 5.2). Como precaução, todos os pacientes do sexo masculino que tomam lenalidomida devem atender às seguintes condições:
• Esteja ciente do risco teratogênico esperado no caso de atividade sexual com uma mulher grávida ou com potencial para engravidar
• Esteja ciente da necessidade de usar preservativos em caso de atividade sexual com uma mulher grávida ou potencialmente fértil que não usa métodos anticoncepcionais eficazes (mesmo se o homem fez vasectomia) durante o tratamento e por 1 semana após a suspensão da dose e / ou tratamento descontinuação.
• Esteja ciente de que se a parceira engravidar enquanto a paciente estiver tomando Revlimid ou logo após interromper o tratamento com Revlimid, deve informar o médico imediatamente e encaminhar a parceira a um especialista ou teratologista que pode avaliar a situação e dar uma opinião.
No caso de mulheres com potencial para engravidar, o médico deve garantir que:
• A paciente atende aos requisitos do Programa de Prevenção de Gravidez, incluindo a confirmação de que ela possui um nível adequado de compreensão
• O paciente aceitou as condições mencionadas acima
Contracepção
Mulheres com potencial para engravidar devem usar anticoncepcionais eficazes por 4 semanas antes da terapia, durante a terapia e até 4 semanas após a terapia com lenalidomida, e também em caso de interrupção da dose, a menos que a paciente se comprometa a observar uma "abstinência absoluta e contínua, mês confirmado Se a terapia anticoncepcional eficaz ainda não tiver sido iniciada, a paciente deve ser encaminhada a um médico especialista para estabelecer um método anticoncepcional eficaz.
Abaixo estão alguns exemplos de métodos anticoncepcionais considerados adequados:
• Planta
• Sistema intrauterino de liberação de levonorgestrel (SIU)
• Depósito de acetato de medroxiprogesterona
• Esterilização tubária
• Relações sexuais apenas com parceiro masculino vasectomizado; vasectomia deve ser confirmada por duas análises de sêmen negativas
• Pílulas só de progestógeno para inibir a ovulação (por exemplo, desogestrel)
Devido ao risco aumentado de tromboembolismo venoso em pacientes com mieloma múltiplo tomando lenalidomida em regimes de combinação, e em menor extensão em pacientes com síndromes mielodisplásicas e linfoma de células do manto tomando lenalidomida sozinha, o uso de contraceptivos orais não é recomendado. Tipo combinado (ver também a seção 4.5). Se a paciente estiver tomando um anticoncepcional oral combinado, ela deve substituir o método anticoncepcional por um dos anteriores. O risco de tromboembolismo venoso permanece por 4-6 semanas após a descontinuação do anticoncepcional oral combinado. A eficácia dos esteróides contraceptivos pode ser reduzida durante o tratamento concomitante com dexametasona (ver secção 4.5).
Implantes e sistemas intrauterinos com liberação de levonorgestrel estão associados a um risco aumentado de infecção na inserção, bem como sangramento vaginal irregular. A profilaxia antibiótica deve ser considerada, principalmente em pacientes com neutropenia.
Dispositivos intrauterinos de liberação de cobre geralmente não são recomendados devido ao risco potencial de infecção no momento da inserção e devido à perda de sangue menstrual, que pode afetar adversamente pacientes com neutropenia ou trombocitopenia.
Teste de gravidez
De acordo com a prática local, em pacientes com potencial para engravidar, testes de gravidez com sensibilidade mínima de 25 mUI / ml devem ser realizados sob supervisão médica, conforme descrito a seguir. Essa obrigação também é válida para pacientes com potencial para engravidar que pratiquem abstinência absoluta e contínua. O ideal é que o teste de gravidez, a prescrição e a dispensa do medicamento ocorram no mesmo dia. A lenalidomida deve ser dispensada a pacientes com potencial para engravidar no prazo de 7 dias a partir da data de prescrição.
Antes de iniciar o tratamento
Quando a paciente estiver usando anticoncepcionais eficazes por pelo menos 4 semanas, um teste de gravidez com supervisão médica deve ser realizado durante a consulta em que a lenalidomida é prescrita ou nos 3 dias anteriores à consulta ao médico. O teste deve garantir que a paciente não está grávida antes de iniciar o tratamento com lenalidomida.
Acompanhamento e fim do tratamento
Um teste de gravidez supervisionado por um médico deve ser repetido a cada 4 semanas, incluindo 4 semanas após o final do tratamento, exceto em casos de esterilização tubária confirmada. Estes testes de gravidez devem ser realizados no mesmo dia da prescrição do médico ou nos 3 dias anteriores à consulta do médico.
Pacientes masculinos
Durante o tratamento, a lenalidomida está presente em níveis extremamente baixos no sêmen e é indetectável no sêmen de indivíduos saudáveis 3 dias após a interrupção do medicamento (ver seção 5.2). Como precaução e tendo em consideração populações especiais de doentes com um tempo de eliminação prolongado, como doentes com compromisso renal, todos os doentes do sexo masculino a tomar lenalidomida devem usar preservativos durante toda a duração do tratamento, durante a descontinuação do tratamento. Dose e até uma semana após a interrupção terapêutica, se a sua parceira estiver grávida ou com potencial para engravidar e não estiver a utilizar métodos contracetivos eficazes (mesmo que o homem tenha feito uma vasectomia).
Precauções adicionais de uso
Os doentes devem ser instruídos a nunca dar este medicamento a outras pessoas e a devolver as cápsulas não utilizadas ao farmacêutico no final do tratamento.
Os pacientes não devem doar sangue durante a terapia com lenalidomida e por pelo menos uma semana após a interrupção do tratamento.
Material educacional, prescrição e restrições de dispensação
Para ajudar os pacientes a evitar a exposição fetal à lenalidomida, o Titular da Autorização de Introdução no Mercado fornecerá materiais educacionais ao pessoal médico para reforçar as advertências sobre a teratogenicidade esperada da lenalidomida, aconselhar sobre contracepção antes do início da terapia e fornecer orientações sobre a necessidade de um teste de gravidez . O médico deve informar as pacientes do sexo masculino e feminino sobre o risco teratogênico e as medidas rígidas de prevenção da gravidez, conforme especificado no Programa de Prevenção da Gravidez, e fornecer às pacientes a cartilha educativa adequada, cartão do paciente e / ou instrumento equivalente, de acordo com as medidas implementadas em a nível nacional. Foi implementado um sistema nacional de controlo da distribuição em colaboração com cada Autoridade Nacional Competente. Este sistema prevê a utilização de um cartão de doente e / ou instrumento equivalente, para o controlo da prescrição e dispensação, e a recolha de dados detalhados relativos à indicação, a fim de controlar cuidadosamente o uso off-label do medicamento em território nacional. Idealmente, o teste de gravidez, a emissão da receita e a dispensa do medicamento devem ocorrer no mesmo dia. A lenalidomida deve ser dispensada a pacientes com potencial para engravidar no prazo de 7 dias a contar da data da receita e após o "resultado negativo do teste de gravidez realizado sob supervisão médica.
Avisos e precauções especiais adicionais de uso
Doenças Cardiovasculares
Infarto do miocárdio
Foram observados casos de enfarte do miocárdio em doentes que receberam lenalidomida, particularmente naqueles com factores de risco conhecidos, e nos primeiros 12 meses quando administrado em combinação com dexametasona. Pacientes com fatores de risco conhecidos, incluindo aqueles com trombose anterior, devem ser monitorados de perto e ações devem ser tomadas para tentar minimizar todos os fatores de risco modificáveis (por exemplo, tabagismo, hipertensão e hiperlipidemia).
Eventos tromboembólicos venosos e arteriais
Em pacientes com mieloma múltiplo, a combinação de lenalidomida e dexametasona está associada a um risco aumentado de tromboembolismo venoso (principalmente trombose venosa profunda e embolia pulmonar) e tromboembolismo arterial (principalmente infarto do miocárdio e evento cerebrovascular). Tromboembolismo venoso foi observado em menor grau. extensão com lenalidomida em combinação com melfalano e prednisona em mieloma múltiplo recentemente diagnosticado e como monoterapia em síndromes mielodisplásicas Ver secções 4.5 e 4.8.
Em pacientes com síndromes mielodisplásicas e linfoma de células do manto, o tratamento com lenalidomida isolada também foi associado a um risco de tromboembolismo venoso (principalmente trombose venosa profunda e embolia pulmonar), mas em menor extensão do que em pacientes com mieloma múltiplo - ver seções 4.5 e 4,8.
Portanto, os pacientes com fatores de risco conhecidos para tromboembolismo - incluindo uma trombose prévia - devem ser monitorados de perto. Ações devem ser tomadas para tentar minimizar todos os fatores de risco modificáveis (por exemplo, tabagismo, hipertensão e hiperlipidemia). Nesses pacientes, a administração concomitante de agentes eritropoiéticos ou uma história prévia de eventos tromboembólicos também podem aumentar o risco de trombose. Portanto, é recomendado, em pacientes com mieloma múltiplo tomando lenalidomida e dexametasona, que os agentes eritropoiéticos ou outros agentes que podem aumentar o risco de trombose, como por exemplo. terapia de reposição hormonal. Se a concentração de hemoglobina aumentar além de 12 g / dl, o uso de agentes eritropoiéticos deve ser interrompido.
Pacientes e médicos devem estar cientes da necessidade de prestar atenção aos sinais e sintomas de tromboembolismo. Os pacientes devem procurar atendimento médico se ocorrerem sintomas como falta de ar, dor no peito, inchaço dos membros superiores ou inferiores. Para fins profiláticos, a ingestão de medicamentos antitrombóticos deve ser recomendada, especialmente em pacientes com fatores de risco trombóticos adicionais.A decisão de adotar medidas antitrombóticas profiláticas deve ser feita após consideração cuidadosa dos fatores de risco para cada paciente individual.
Se o paciente apresentar algum evento tromboembólico, o tratamento deve ser interrompido e iniciada a terapia anticoagulante padrão. Uma vez que o paciente esteja estabilizado com anticoagulação e todas as complicações do evento tromboembólico tenham sido resolvidas, o tratamento com lenalidomida pode ser reiniciado com a dose original após uma avaliação risco-benefício.O paciente deve continuar a terapia de anticoagulação durante o tratamento.
Neutropenia e trombocitopenia
As principais toxicidades limitantes da dose da lenalidomida incluem neutropenia e trombocitopenia. A fim de monitorar a possível ocorrência de citopenia, uma contagem completa de células sanguíneas, incluindo contagem de leucócitos incluindo diferencial, contagem de plaquetas, hemoglobina e hematócrito, no início do estudo, uma vez por semana, deve ser realizada durante as primeiras 8 semanas de tratamento. lenalidomida e uma vez por mês depois disso.Em pacientes com linfoma de células do manto, o cronograma de monitoramento deve ser a cada 2 semanas nos ciclos 3 e 4 e no início de cada ciclo subsequente. Pode ser necessária uma redução da dose (ver secção 4.2). Em caso de neutropenia, o médico deve considerar o uso de fatores de crescimento no tratamento do paciente. Os pacientes devem ser aconselhados a relatar episódios febris imediatamente. Aconselha-se precaução na administração concomitante de lenalidomida com outros agentes mielossupressores.
• Mieloma múltiplo recém-diagnosticado em pacientes tratados com lenalidomida em combinação com dexametasona em dose baixa
Neutropenia de grau 4 foi observada em menor extensão na lenalidomida em combinação com braços de tratamento de dexametasona em dose baixa em comparação com o braço comparador (8,5% em Rd [tratamento contínuo] e Rd18 [tratamento por 18 ciclos de quatro semanas], em comparação com 15 % no braço do melfalano / prednisona / talidomida, ver secção 4.8). Os episódios de neutropenia febril de grau 4 foram consistentes com o braço comparador (0,6% em doentes tratados com lenalidomida / dexametasona Rd e Rd18, em comparação com 0,7% em doentes no braço melfalano / prednisona / talidomida, ver secção 4.8). Os doentes devem ser aconselhados a notificar imediatamente episódios febris e pode ser necessária redução da dose (ver secção 4.2).
Trombocitopenia de grau 3 ou 4 foi observada em menor extensão nos braços Rd e Rd18 do que no braço comparador (8,1% vs 11,1%, respectivamente). Os doentes e médicos devem observar os sinais e sintomas de hemorragia, incluindo petéquias e epistaxe, especialmente em doentes submetidos a tratamento concomitante que possa induzir hemorragia (ver secção 4.8, Distúrbios hemorrágicos).
• Mieloma múltiplo recém-diagnosticado em pacientes tratados com lenalidomida em combinação com melfalano e prednisona
Em estudos clínicos em pacientes com mieloma múltiplo recém-diagnosticado, a combinação de lenalidomida com melfalano e prednisona está associada a uma maior incidência de neutropenia de grau 4 (34,1% em pacientes no braço de melfalano, prednisona e lenalidomida seguido por lenalidomida [MPR + R] e melfalano, prednisona e lenalidomida seguidos de placebo [MPR + p], em comparação com 7,8% dos pacientes tratados com MPp + p; ver seção 4.8). Episódios de neutropenia febril de grau 4 não foram observados com frequência (1,7% em pacientes tratados com MPR + R / MPR + p, em comparação com 0,0% em doentes tratados com MPp + p; ver secção 4.8).
Em pacientes com mieloma múltiplo, a combinação de lenalidomida com melfalano e prednisona está associada a uma maior incidência de trombocitopenia de grau 3 e grau 4 (40,4% em pacientes tratados com MMR + R / MMR + p, em comparação com 13,7% em pacientes tratados com MPp + p; ver secção 4.8) Os doentes e médicos devem estar alertas para sinais e sintomas de hemorragia, incluindo petéquias e epistaxe, especialmente em doentes tratados concomitantemente com medicamentos que aumentam a predisposição à hemorragia (ver secção 4.8, Alterações hemorrágicas).
• Mieloma múltiplo com pelo menos uma terapia anterior
Em pacientes com mieloma múltiplo recebendo pelo menos uma terapia anterior, a combinação de lenalidomida e dexametasona está associada a uma maior incidência de neutropenia de grau 4 (5,1% dos pacientes tratados com lenalidomida / dexametasona em comparação com 0,6% dos pacientes tratados com placebo / dexametasona; ver secção 4.8). Episódios de neutropenia febril de grau 4 foram observados com pouca frequência (em 0,6% dos doentes tratados com lenalidomida / dexametasona em comparação com 0,0% dos doentes tratados com placebo / dexametasona; ver secção 4.8) Os doentes devem ser aconselhados a notificar imediatamente episódios febris Pode ser necessária redução da dose (ver secção 4.2) Em caso de neutropenia, os médicos devem considerar a utilização de fatores de crescimento no tratamento do doente.
Em pacientes com mieloma múltiplo, a combinação de lenalidomida e dexametasona está associada a uma maior incidência de trombocitopenia de grau 3 e 4 (9,9% e 1,4%, respectivamente, a maior incidência de trombocitopenia de grau 3). 3 e Grau 4 (9,9%) e 1,4% dos pacientes tratados com lenalidomida / dexametasona, respectivamente, em comparação com 2,3% e 0,0% dos pacientes tratados com placebo / dexametasona; ver seção 4.8). Pacientes e médicos devem monitorar sinais e sintomas de sangramento, incluindo petéquias e epistaxe, especialmente em pacientes tratados concomitantemente com medicamentos que podem induzir hemorragia (ver secção 4.8, Distúrbios hemorrágicos).
• Síndromes mielodisplásicas
Em doentes com síndromes mielodisplásicas, o tratamento com lenalidomida está associado a uma maior incidência de neutropenia e trombocitopenia de grau 3 e 4 do que em doentes tratados com placebo (ver secção 4.8).
• Linfoma de células do manto
Em doentes com linfoma de células do manto, o tratamento com lenalidomida está associado a uma maior incidência de neutropenia de grau 3 e 4 do que em doentes no braço de controlo (ver secção 4.8).
Infecção com ou sem neutropenia
Pacientes com mieloma múltiplo são propensos a desenvolver infecções, incluindo pneumonia. Foi observada uma taxa mais elevada de infecções durante o tratamento com lenalidomida em combinação com dexametasona do que com MPT. Infecções de grau ≥ 3 ocorreram no contexto de neutropenia em menos de um terço dos pacientes. Pacientes com fatores de risco conhecidos para infecções devem ser monitorados de perto. Todos os pacientes devem ser aconselhados a consultar seu médico imediatamente ao primeiro sinal de infecção (por exemplo, tosse, febre, etc.), para permitir uma intervenção imediata para reduzir a gravidade.
Falência renal
A lenalidomida é substancialmente excretada pelos rins. Assim, em doentes com insuficiência renal, deve ter-se um cuidado especial na escolha da dose e é aconselhável a monitorização da função renal (ver secção 4.2).
Distúrbios da glândula tireóide
Foram observados casos de hipotireoidismo e hipertireoidismo. Antes de iniciar o tratamento, é recomendado o controle ideal das comorbidades que afetam a função tireoidiana. Recomenda-se que a função tireoidiana seja monitorada no início e durante o tratamento.
Neuropatia periférica
A lenalidomida está estruturalmente relacionada à talidomida, que é conhecida por causar neuropatia periférica grave. Não houve aumento na neuropatia periférica observada com o uso de lenalidomida para o tratamento de mieloma múltiplo recém-diagnosticado.
Reação de exacerbação tumoral e síndrome de lise tumoral
Uma vez que a lenalidomida exibe atividade antineoplásica, complicações da síndrome de lise tumoral (Síndrome de Lise Tumoral, TLS). TLS e o Reação de alargamento de tumor (TFR) foram comumente observados em pacientes com leucemia linfocítica crônica (LLC) e raramente observados em pacientes com linfomas tratados com lenalidomida. Casos de SLT com desfecho fatal foram relatados durante o tratamento com lenalidomida. Os pacientes em risco de TLS e TFR são aqueles com alta carga tumoral antes do tratamento. Deve-se ter cuidado ao iniciar o tratamento com lenalidomida nesses pacientes. Recomenda-se que tais pacientes sejam monitorados cuidadosamente, especialmente durante o primeiro ciclo ou aumento da dose, e que as precauções apropriadas sejam tomadas. Houve relatos raros de SLT em pacientes com MM tratados com lenalidomida, enquanto nenhum caso foi relatado em pacientes com MDS tratado com lenalidomida.
Massa tumoral
• Linfoma de células do manto
A lenalidomida não é recomendada para o tratamento de pacientes com alta carga tumoral se houver opções de tratamento alternativas disponíveis.
Morte precoce
No Estudo MCL-002, houve um aumento geral evidente nas mortes prematuras (dentro de 20 semanas). Pacientes com alta carga tumoral basal têm um risco maior de morte precoce: houve 16/81 (20%) mortes prematuras no braço da lenalidomida e 2/28 (7%) mortes prematuras no braço controle. Em 52 semanas, os números correspondentes eram 32/81 (40%) e 6/28 (21%) (ver seção 5.1).
Eventos adversos
No Estudo MCL-002 durante o ciclo de tratamento 1, 11/81 (14%) pacientes com alta carga tumoral foram retirados da terapia com lenalidomida, em comparação com 1/28 (4%) no grupo de controle. A principal razão para descontinuar o tratamento para pacientes com alta carga tumoral durante o ciclo de tratamento 1 no braço da lenalidomida foi devido a eventos adversos, 7/11 (64%).
Pacientes com alta carga tumoral devem, portanto, ser cuidadosamente monitorados quanto a reações adversas (ver seção 4.8), incluindo quaisquer sinais de Reação de alargamento de tumor (TFR). Para ajustes de dose em caso de TFR, ver seção 4.2.
Uma massa tumoral elevada foi definida como pelo menos uma lesão ≥ 5 cm de diâmetro ou 3 lesões ≥ 3 cm.
Reação de alargamento de tumor
• Linfoma de células do manto
Recomenda-se monitoramento e avaliação cuidadosos para TFR. Pacientes com MIPI elevada (Índice Prognóstico Internacional de Linfoma de Células de Mantel) no momento do diagnóstico ou doença caracterizada por grandes massas tumorais (pelo menos uma lesão ≥ 7 cm no diâmetro mais longo) no início do estudo pode estar em risco de TFR. Lá Reação de alargamento de tumor pode simular a progressão da doença (DP). Os pacientes nos estudos MCL-002 e MCL-001 que apresentaram TFR de grau 1 e 2 foram tratados com corticosteroides, antiinflamatórios não esteroidais (AINEs) e / ou analgésicos narcóticos para o manejo dos sintomas de TFR. A decisão de adotar medidas terapêuticas para TFR deve ser feita após uma “avaliação clínica cuidadosa de cada paciente (ver seção 4.2).
Reações alérgicas
Foram notificados casos de reações alérgicas / de hipersensibilidade em doentes tratados com lenalidomida (ver secção 4.8). Recomenda-se monitorar cuidadosamente os pacientes que tiveram reações alérgicas anteriores à talidomida, uma vez que uma possível reação cruzada entre lenalidomida e talidomida foi relatada na literatura.
Reações cutâneas graves
Casos de SSJ e NET foram relatados. O tratamento com lenalidomida deve ser interrompido no caso de erupção cutânea esfoliativa ou bolhosa, ou se houver suspeita de SSJ ou NET, e não deve ser retomado após a interrupção devido a essas reações. A interrupção ou descontinuação da lenalidomida deve ser considerada para outras formas de reações cutâneas, dependendo de sua gravidade.Os pacientes com história prévia de erupção cutânea grave associada ao tratamento com talidomida não devem receber lenalidomida.
Intolerância a lactose
As cápsulas de Revlimid contêm lactose. Os doentes com problemas hereditários raros de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glucose-galactose não devem tomar este medicamento.
Cápsulas não utilizadas
Os doentes devem ser aconselhados a nunca dar este medicamento a outras pessoas e a devolver as cápsulas não utilizadas ao farmacêutico no final do tratamento.
Segundo tumor primário
Um aumento nos segundos tumores primários (Segundo Primário Malignidade, SPM) em pacientes com mieloma previamente tratados com lenalidomida / dexametasona (3,98 por 100 pessoas-ano) versus controles (1,38 por 100 pessoas-ano). Os SPMs não invasivos consistem em carcinomas basocelulares ou espinocelulares.
A maioria dos SPMs invasivos eram tumores sólidos.
Em estudos clínicos em pacientes recém-diagnosticados com mieloma múltiplo inelegíveis para transplante, um aumento de 4,9 vezes na taxa de incidência de PMS hematológica (casos de AML, MDS) foi observado em pacientes tratados com lenalidomida em combinação com melfalano e prednisona para progressão (1,75 por 100 pessoas-ano), em comparação com melfalano em combinação com prednisona (0,36 por 100 por pessoa-ano).
Um aumento de 2,12 vezes na taxa de incidência de SPM sólido foi observado em pacientes tratados com lenalidomida (9 ciclos) em combinação com melfalano e prednisona (1,57 por 100 pessoas-ano), em comparação com melfalano em combinação com prednisona (0,74 por 100 por pessoa-anos).
Em pacientes tratados com lenalidomida em combinação com dexametasona até a progressão ou por 18 meses, a taxa de incidência de PMS hematológica (0,16 por 100 pessoas-ano) não foi aumentada em comparação com a talidomida em combinação com melfalano e prednisona (0,79 por 100 pessoas-ano) .
Um aumento de 1,3 vezes na taxa de incidência de PMS sólido foi observado em pacientes tratados com lenalidomida em combinação com dexametasona até a progressão ou por 18 meses (1,58 por 100 pessoas-ano) em comparação com a talidomida em combinação com melfalano e prednisona (1,19 por 100 pessoa-anos).
Em ensaios clínicos em pacientes recém-diagnosticados com mieloma múltiplo elegíveis para transplante, uma taxa de incidência aumentada de PMS hematológica foi observada em pacientes tratados com lenalidomida imediatamente após transplante de melfalano em alta dose e células-tronco autólogas (Transplante autólogo de células-tronco, ASCT), em comparação com pacientes tratados com placebo (1,27 a 1,56 e 0,46 a 0,53 por 100 pessoas-ano, respectivamente). Os casos de tumores malignos de células B (incluindo linfoma de Hodgkin) observados em ensaios clínicos ocorreram em pacientes tratados com lenalidomida no contexto pós-ASCT.
O risco de PMS hematológica deve ser considerado antes de iniciar o tratamento com Revlimid em associação com melfalano ou no período imediatamente a seguir a uma dose elevada de melfalano e ASCT. Os médicos devem avaliar cuidadosamente os pacientes antes e durante o tratamento, usando o rastreamento padrão do câncer para TPM e instituir o tratamento conforme as instruções.
Progressão para leucemia mieloide aguda (LMA) na síndrome mielodisplásica (SMD) em risco baixo ou intermediário-1
• Cariótipo
Variáveis basais, incluindo anormalidades citogenéticas complexas, estão associadas à progressão para LMA em indivíduos dependentes de transfusão com anormalidade de deleção 5q isolada. Em uma análise combinada de dois estudos clínicos conduzidos com Revlimid em SMD de risco baixo ou intermediário-1, os indivíduos com anormalidades citogenéticas complexas tiveram o risco cumulativo mais alto de progressão para LMA estimado em 2 anos (38,6%). Taxa de progressão estimada em 2 anos. para AML em pacientes com anormalidade de deleção 5q isolada foi de 13,8%, em comparação com 17,3% para pacientes com anormalidade de deleção 5q isolada e uma "anormalidade citogenética adicional.
Consequentemente, a relação benefício / risco de Revlimid é desconhecida quando a SMD está associada a anomalias de deleção 5q isoladas e anomalias citogenéticas complexas.
• Status TP53
Uma mutação TP53 está presente em 20-25% dos pacientes com SMD com anomalia de deleção isolada 5q de baixo risco e está associada a um maior risco de progressão para LMA. Em uma "análise post-hoc de um estudo clínico (MDS-004) conduzido com Revlimid em MDS de risco baixo ou intermediário-1, a taxa de progressão estimada de 2 anos para LMA foi de 27,5% em pacientes com IHC-p53 positivo (1 % cut-off de coloração nuclear forte, usando avaliação imunohistoquímica da proteína p53 como substituta para o estado de mutação TP53) e 3,6% em pacientes com IHC-p53 negativo (p = 0,0038) (ver seção 4.8).
Progressão para outras doenças malignas no linfoma de células do manto
No linfoma de células do manto, LMA, tumores malignos de células B e câncer de pele não melanoma (CPNM) são riscos potenciais.
Distúrbios hepáticos
Casos de insuficiência hepática, incluindo com desfecho fatal, foram observados em pacientes tratados com lenalidomida em terapia combinada: insuficiência hepática aguda, hepatite tóxica, hepatite citolítica, hepatite colestática e hepatite citolítica / colestática mista. Os mecanismos de hepatotoxicidade grave induzida por medicamentos permanecem desconhecidos, embora, em alguns casos, os fatores de risco possam ser doença hepática viral pré-existente, enzimas hepáticas basais elevadas e possivelmente tratamento com antibióticos.
Anormalidades nos testes de função hepática foram comumente observadas e geralmente eram assintomáticas e reversíveis com a descontinuação do tratamento. Uma vez que os parâmetros tenham retornado aos valores basais, pode-se considerar a retomada do tratamento com uma dose mais baixa.
A lenalidomida é excretada pelos rins. É importante ajustar a dose em doentes com insuficiência renal para evitar atingir os níveis plasmáticos que podem aumentar o risco de reações adversas hematológicas mais importantes ou hepatotoxicidade. Recomenda-se a monitorização da função hepática, particularmente no caso de infecção hepática viral anterior ou concomitante ou quando a lenalidomida é administrada em combinação com medicamentos conhecidos por estarem associados a disfunção hepática.
Pacientes com mieloma múltiplo recém-diagnosticado
Houve uma maior taxa de intolerância (eventos adversos de grau 3 ou 4, eventos adversos graves, descontinuação do tratamento) em pacientes com idade> 75 anos, estágio ISS (Sistema Internacional de Estadiamento) III, status de desempenho (PS) ≤ 2 avaliado de acordo com os critérios ECOG (Grupo de Oncologia Cooperativa Oriental) ou CLcr
Catarata
Cataratas foram observadas com mais frequência em pacientes tratados com lenalidomida em combinação com dexametasona, particularmente quando usada por um período prolongado. Recomenda-se monitorar periodicamente a capacidade visual.
04.5 Interações com outros medicamentos e outras formas de interação -
Os agentes eritropoiéticos, ou outros agentes que podem aumentar o risco de trombose, como a terapia de reposição hormonal, devem ser usados com cautela em pacientes com mieloma múltiplo tomando lenalidomida e dexametasona (ver seções 4.4 e 4.8). tomar lenalidomida e dexametasona (ver secções 4.4 e 4.8).
Contraceptivos orais
Não foram realizados estudos de interação com contraceptivos orais. A lenalidomida não é um indutor enzimático. Em um estúdio em vitro conduzida com hepatócitos humanos, lenalidomida, testada em várias concentrações, não induziu CYP1A2, CYP2B6, CYP2C9, CYP2C19 e CYP3A4 / 5. Portanto, se a lenalidomida for administrada isoladamente, não é esperada indução que leve à redução da eficácia dos medicamentos, incluindo contraceptivos hormonais. No entanto, a dexametasona é conhecida por ser um indutor fraco a moderado do CYP3A4 e provavelmente afeta outras enzimas e proteínas de transporte. Não está excluído que a eficácia dos contraceptivos orais pode ser reduzida durante o tratamento.
Devem ser tomadas medidas eficazes para evitar a gravidez (ver secções 4.4 e 4.6).
Varfarina
A administração concomitante de 10 mg de doses repetidas de lenalidomida não teve efeito na farmacocinética de dose única de R- e S-varfarina. A co-administração de uma dose única de 25 mg de varfarina não teve efeito na farmacocinética da lenalidomida. No entanto, não se sabe se existe uma "interação durante" o uso clínico (tratamento concomitante com dexametasona). A dexametasona é um indutor enzimático fraco a moderado e seus efeitos sobre a varfarina são desconhecidos. É aconselhável monitorar de perto a concentração de varfarina durante o tratamento.
Digoxina
A administração concomitante de 10 mg / dia de lenalidomida aumentou a concentração plasmática de digoxina em 14% (0,5 mg, dose única) com um IC (intervalo de confiança) de 90% [0,52% -28,2%]. Não se sabe se o efeito seria diferente na situação terapêutica (doses mais elevadas de lenalidomida e tratamento concomitante com dexametasona) .Portanto, a monitorização da concentração de digoxina é recomendada durante o tratamento com lenalidomida.
Estatinas
Quando as estatinas são administradas com lenalidomida, há um risco aumentado de rabdomiólise, que pode ser simplesmente aditiva. O monitoramento clínico e laboratorial aprimorado é garantido, principalmente durante as primeiras semanas de tratamento.
Dexametasona
A co-administração de doses únicas ou múltiplas de dexametasona (40 mg / dia) não tem efeito clinicamente relevante na farmacocinética de doses múltiplas da lenalidomida (25 mg / dia).
Interações com inibidores da glicoproteína P (gp-P)
Em vitro, A lenalidomida é um substrato da P-gp, mas não é um inibidor da P-gp. A co-administração de doses múltiplas do inibidor potente da gp-P, quinidina (600 mg, duas vezes ao dia) ou do inibidor / substrato da gp-P de ação moderada temsirolímus (25 mg), não tem efeito clinicamente relevante na farmacocinética da lenalidomida (25 mg) A administração concomitante de lenalidomida não altera a farmacocinética do temsirolímus.
04.6 Gravidez e amamentação -
Mulheres com potencial para engravidar / Contracepção em homens e mulheres
Mulheres com potencial para engravidar devem usar métodos anticoncepcionais eficazes. Se ocorrer gravidez durante o tratamento com lenalidomida, a terapia deve ser interrompida e a paciente deve ir a um especialista ou com experiência em teratologia que possa avaliar a situação e dar uma opinião. Se o parceiro de um paciente do sexo masculino em tratamento com lenalidomida estiver grávida, o parceiro deve ser aconselhado a ir a um médico especialista ou a um médico com experiência em teratologia que possa avaliar a situação e dar uma opinião.
Durante o tratamento, a lenalidomida está presente em níveis extremamente baixos no sêmen e é indetectável no sêmen de indivíduos saudáveis 3 dias após a interrupção do medicamento (ver seção 5.2). Por precaução e tendo em consideração populações especiais de doentes com um tempo de eliminação prolongado, como doentes com compromisso renal, todos os doentes do sexo masculino a tomar lenalidomida devem usar preservativos durante toda a duração do tratamento, durante a suspensão da dose e até uma semana após a interrupção do tratamento se sua parceira está grávida ou com potencial para engravidar e não usa nenhum método contraceptivo.
Gravidez
A lenalidomida está estruturalmente relacionada à talidomida, uma substância ativa com conhecido efeito teratogênico em humanos, que causa malformações congênitas graves com risco de vida.
A lenalidomida induziu malformações em macacos semelhantes às descritas para a talidomida (ver secção 5.3). Portanto, é esperado um efeito teratogênico da lenalidomida, e a lenalidomida está contra-indicada durante a gravidez (ver secção 4.3).
Hora da alimentação
Como não se sabe se a lenalidomida é excretada no leite materno, recomenda-se que a amamentação seja interrompida durante a terapia com lenalidomida.
Fertilidade
Um estudo de fertilidade conduzido em ratos com doses de lenalidomida de até 500 mg / kg (aproximadamente 200 a 500 vezes as doses de 25 mg e 10 mg, respectivamente, usadas em humanos e calculadas com base na área de superfície corporal), não mostrou efeitos adversos em fertilidade ou toxicidade materna.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas -
A lenalidomida tem um efeito ligeiro a moderado na capacidade de conduzir e utilizar máquinas. Fadiga, tonturas, sonolência, tonturas e visão turva foram relatados durante o tratamento com lenalidomida. Portanto, recomenda-se cuidado ao dirigir veículos ou usar máquinas.
04.8 Efeitos indesejáveis -
Resumo do perfil de segurança
Recém-diagnosticado mieloma múltiplo em pacientes tratados com lenalidomida em combinação com baixa dose de dexametasona
As reações adversas graves mais frequentemente observadas (≥ 5%) com lenalidomida em combinação com dexametasona em dose baixa (Rd e Rd18), em comparação com melfalano, prednisona e talidomida (MPT), foram:
• Pneumonia (9,8%)
• Insuficiência renal (incluindo aguda) (6,3%)
As reações adversas observadas mais frequentemente com Rd ou Rd18 do que com MPT foram: diarreia (45,5%), fadiga (32,8%), dor nas costas (32,0%), astenia (28,2%), insônia (27,6%), erupção cutânea (24,3%) , diminuição do apetite (23,1%), tosse (22,7%), pirexia (21,4%) e espasmos musculares (20,5%).
Pacientes recém-diagnosticados com mieloma múltiplo tratados com lenalidomida em combinação com melfalano e prednisona
As reações adversas graves mais frequentemente observadas (≥ 5%) com melfalano, prednisona e lenalidomida seguidas de terapia de manutenção com lenalidomida (MPR + R) ou melfalano, prednisona e lenalidomida seguida de placebo (MPR + p), em comparação com melfalano, prednisona e placebo seguido por placebo (MPp + p), foram:
• Neutropenia febril (6,0%)
• Anemia (5,3%)
As reações adversas observadas mais frequentemente com MPR + R ou MPR + p do que com MPp + p foram: neutropenia (83,3%), anemia (70,7%), trombocitopenia (70,0%), leucopenia (38,8%), constipação (34,0 %), diarreia (33,3%), erupção cutânea (28,9%), pirexia (27,0%), edema periférico (25,0%), tosse (24,0%), diminuição do apetite (23,7%) e astenia (22,0%).
Mieloma múltiplo com pelo menos uma terapia anterior
Em dois estudos de Fase III controlados por placebo, 353 pacientes com mieloma múltiplo foram expostos ao tratamento combinado com lenalidomida / dexametasona e 351 ao tratamento combinado com placebo / dexametasona.
As reações adversas mais graves observadas com mais frequência com a combinação de lenalidomida / dexametasona do que com a combinação de placebo / dexametasona foram:
• Tromboembolismo venoso (trombose venosa profunda, embolia pulmonar) (ver seção 4.4)
• Neutropenia de grau 4 (ver secção 4.4).
As reações adversas que ocorreram com mais frequência com lenalidomida e dexametasona, em comparação com placebo e dexametasona, quando combinados com ensaios clínicos de mieloma múltiplo (MM-009 e MM-010), foram fadiga (43,9%), neutropenia (42,2%), constipação ( 40,5%), diarreia (38,5%), cãibras musculares (33,4%), anemia (31,4%), trombocitopenia (21,5%) e erupção cutânea (21,2%).
Síndromes mielodisplásicas
O perfil de segurança geral de Revlimid em doentes com síndromes mielodisplásicas é baseado em dados de um total de 286 doentes incluídos num estudo de Fase II e Fase III (ver secção 5.1). Na Fase II, todos os 148 pacientes estavam em tratamento com lenalidomida. No estudo de Fase III, 69 pacientes foram tratados com lenalidomida 5 mg, 69 pacientes foram tratados com lenalidomida 10 mg e 67 pacientes receberam placebo durante a fase duplo-cega do estudo.
A maioria das reações adversas tendeu a ocorrer durante as primeiras 16 semanas de terapia com lenalidomida.
As reações adversas graves incluem:
• Tromboembolismo venoso (trombose venosa profunda, embolia pulmonar) (ver seção 4.4)
• Neutropenia de grau 3 ou 4, neutropenia febril e trombocitopenia de grau 3 ou 4 (ver secção 4.4).
As reações adversas mais comumente observadas que ocorreram com mais frequência nos grupos de lenalidomida do que no braço de controle (placebo) no estudo de Fase III foram neutropenia, (76,8%), trombocitopenia (46,4%)., Diarreia (34,8%), prisão de ventre ( 19,6%), náuseas (19,6%), prurido (25,4%), erupção cutânea (18,1%), fadiga (18,1%) e espasmos musculares (16,7%).
Linfoma de células do manto
O perfil de segurança geral de Revlimid em doentes com linfoma de células do manto é baseado em dados de 254 doentes incluídos num estudo de Fase II aleatório e controlado, MCL-002 (ver secção 5.1).
Além disso, as reações adversas a medicamentos (ADRs) observadas no estudo de apoio MCL-001 foram incluídas na Tabela 3.
As reações adversas graves observadas com mais frequência no Estudo MCL-002 (com uma diferença de pelo menos 2 pontos percentuais) no braço da lenalidomida versus o braço de controle foram:
• Neutropenia (3,6%)
• Embolia pulmonar (3,6%)
• Diarreia (3,6%)
As reações adversas mais comumente observadas ocorrendo com mais frequência no braço da lenalidomida do que no braço controle no Estudo MCL-002 foram neutropenia, (50,9%), anemia (28,7%), diarreia (22,8%), fadiga (21,0%) , constipação (17,4%), pirexia (16,8%) e erupção cutânea (incluindo dermatite alérgica) (16,2%).
No Estudo MCL-002, houve um aumento geral evidente nas mortes prematuras (dentro de 20 semanas). Pacientes com alta carga tumoral basal têm um risco maior de morte precoce: 16/81 (20%) mortes prematuras no braço da lenalidomida e 2/28 (7%) mortes prematuras no braço controle. Em 52 semanas, os números correspondentes eram 32/81 (39,5%) e 6/28 (21%) (ver seção 5.1).
Durante o ciclo de tratamento 1, 11/81 (14%) pacientes com alta carga tumoral foram retirados da terapia com lenalidomida, em comparação com 1/28 (4%) no grupo de controle. A principal razão para descontinuar o tratamento para pacientes com alta carga tumoral durante o ciclo de tratamento 1 no braço da lenalidomida foi devido a eventos adversos, 7/11 (64%).
Uma massa tumoral elevada foi definida como pelo menos uma lesão ≥ 5 cm de diâmetro ou 3 lesões ≥ 3 cm.
Lista resumida de reações adversas
Tabela de resumo para terapia combinada
As reações adversas observadas em pacientes tratados para mieloma múltiplo estão listadas abaixo por classes de sistemas de órgãos e frequência. Dentro de cada classe de frequência, as reações adversas são listadas por ordem decrescente de gravidade. As frequências são definidas como se segue: muito frequentes (≥ 1/10); frequentes (≥ 1/100,
A tabela a seguir foi compilada com base em dados coletados durante estudos de mieloma múltiplo com terapia combinada. Os dados não foram atualizados tendo em conta a maior duração do tratamento nos braços contendo lenalidomida até à progressão da doença, em comparação com os braços comparadores nos estudos principais de mieloma múltiplo (ver secção 5.1).
As reações adversas foram classificadas na categoria apropriada na tabela abaixo, de acordo com a incidência mais elevada observada em qualquer um dos estudos clínicos principais.
Tabela 1: Reações adversas relatadas em ensaios clínicos em pacientes com mieloma múltiplo tratados com lenalidomida em combinação com dexametasona ou com melfalano e prednisona
^ Consulte a seção 4.8 Descrição de reações adversas selecionadas
* O câncer de pele de células escamosas foi observado em ensaios clínicos em pacientes com mieloma previamente tratados com lenalidomida / dexametasona em comparação com controles
** Câncer de pele de células escamosas foi observado em um estudo clínico em pacientes recém-diagnosticados com mieloma com lenalidomida / dexametasona em comparação com controles
Tabela de resumo para monoterapia
As reações adversas observadas em pacientes tratados para síndromes mielodisplásicas e linfoma de células do manto estão listadas abaixo por classes de sistemas de órgãos e frequência.
Em cada classe de frequência, as reações adversas estão listadas por ordem decrescente de gravidade. A frequência é definida como: muito frequentes (≥ 1/10); frequentes (≥ 1/100,
As tabelas a seguir foram compiladas com base nos dados coletados durante os principais estudos de monoterapia para síndromes mielodisplásicas e linfoma de células do manto.
As reações adversas foram colocadas na categoria apropriada nas tabelas seguintes, de acordo com a incidência mais elevada observada em qualquer um dos estudos clínicos principais.
Tabela 2: Reações adversas relatadas em ensaios clínicos em pacientes com síndromes mielodisplásicas tratados com lenalidomida
^ Consulte a seção 4.8 Descrição de reações adversas selecionadas
♦ Eventos adversos observados como graves em ensaios clínicos de síndromes mielodisplásicas.
≈ Alteração de humor foi observada como um evento adverso sério comum no estudo de síndromes mielodisplásicas de Fase III; não foi relatado como um evento adverso de grau 3 ou 4.
Algoritmo aplicado para a entrada do RCM: Todas as reações adversas a medicamentos (ADRs) capturadas pelo algoritmo de estudo de Fase III estão incluídas no RCM europeu. Para esses ADRs, um controle adicional da frequência de ADRs adquiridos pelo algoritmo de estudo de Fase II foi realizado e, se a frequência de ADRs no estudo de Fase II foi maior do que a registrada no estudo de Fase III, o evento foi incluído em a RCP europeia na frequência observada no estudo de Fase II.
Algoritmo aplicado para síndromes mielodisplásicas:
• Estudo de fase III em síndromes mielodisplásicas (população de segurança duplo-cega, diferença entre lenalidomida 5/10 mg e placebo para o regime de dosagem inicial com ocorrência em pelo menos 2 indivíduos))
o Todos os eventos adversos que ocorrem durante o tratamento em ≥ 5% dos indivíduos tratados com lenalidomida e uma diferença de pelo menos 2% na porcentagem entre lenalidomida e placebo
o Todos os eventos adversos de Grau 3 ou 4 que ocorrem durante o tratamento em 1% dos indivíduos tratados com lenalidomida e uma diferença de pelo menos 1% na porcentagem entre lenalidomida e placebo
o Todos os eventos adversos graves que ocorrem durante o tratamento em 1% dos indivíduos tratados com lenalidomida e uma diferença de pelo menos 1% na porcentagem entre lenalidomida e placebo
• Estudo de fase II em síndromes mielodisplásicas
o Todos os eventos adversos que ocorrem durante o tratamento em ≥ 5% dos indivíduos tratados com lenalidomida o Todos os eventos adversos de grau 3 ou 4 que ocorrem durante o tratamento em 1% dos indivíduos tratados com lenalidomida o Todos os eventos adversos graves que ocorrem durante o tratamento em 1% dos indivíduos tratados com lenalidomida
Tabela 3: Reações adversas relatadas em ensaios clínicos em pacientes com linfoma de células do manto tratados com lenalidomida
^ Consulte a seção 4.8 Descrição de reações adversas selecionadas
♦ Eventos adversos observados como graves em estudos clínicos de linfoma de células do manto.
Algoritmo aplicado para linfoma de células do manto:
• Estudo controlado de fase II em linfoma de células do manto
o Todos os eventos adversos que ocorrem durante o tratamento em ≥ 5% dos indivíduos no braço da lenalidomida e uma diferença de pelo menos 2% na porcentagem entre a lenalidomida e o braço de controle
o Todos os eventos adversos de Grau 3 ou 4 que ocorrem durante o tratamento em ≥ 1% dos indivíduos no braço da lenalidomida e uma diferença de pelo menos 1,0% na porcentagem entre a lenalidomida e o braço controle
o Todos os eventos adversos graves que ocorrem durante o tratamento em ≥ 1% dos indivíduos no braço da lenalidomida e uma diferença de pelo menos 1,0% na porcentagem entre a lenalidomida e o braço controle
• Estudo de um braço de fase II em linfoma de células do manto
o Todos os eventos adversos que ocorrem durante o tratamento em ≥ 5% dos indivíduos
o Todos os eventos adversos de Grau 3 ou 4 que ocorrem durante o tratamento relatados em 2 ou mais indivíduos
Quadro de resumo das reações adversas pós-comercialização
Além das reações adversas acima mencionadas, identificadas nos estudos clínicos essenciais, a seguinte tabela foi compilada com base nos dados coletados no pós-comercialização.
Tabela 4: Reações adversas relatadas no uso pós-comercialização em pacientes tratados com lenalidomida
^ Consulte a seção 4.8 Descrição de reações adversas selecionadas
Descrição das reações adversas selecionadas
Teratogenicidade
A lenalidomida está estruturalmente relacionada com a talidomida, uma substância ativa com conhecido efeito teratogênico em humanos, que causa malformações congênitas graves com risco de vida. Malformações induzidas pela lenalidomida em macacos semelhantes às descritas para a talidomida (ver seções 4.6 e 5.3). Um efeito teratogênico da lenalidomida é esperado em humanos durante a gravidez.
Neutropenia e trombocitopenia
• Pacientes recém-diagnosticados com mieloma múltiplo tratados com lenalidomida em combinação com dexametasona em baixa dose
Em pacientes com mieloma múltiplo recém-diagnosticado, a combinação de lenalidomida com dexametasona em dose baixa está associada a uma incidência reduzida de neutropenia de grau 4 (8,5% em Rd e Rd18, em comparação com 15% em MPT). Neutropenia febril de grau 4 foi observada com pouca frequência (0,6%, em comparação com 0,7% no MPT).
Em pacientes com mieloma múltiplo recém-diagnosticado, a combinação de lenalidomida com dexametasona em dose baixa está associada a uma incidência reduzida de trombocitopenia de grau 3 e 4 (8,1% em Rd e Rd18, em comparação com 11% em MPT).
• Pacientes recém-diagnosticados com mieloma múltiplo tratados com lenalidomida em combinação com melfalano e prednisona
Em pacientes com mieloma múltiplo recém-diagnosticado, a combinação de lenalidomida com melfalano e prednisona está associada a uma maior incidência de neutropenia de grau 4 (34,1% em MMR + R / MPR + p, em comparação com 7,8% em MPp + p). de neutropenia febril de grau 4 (1,7% no MPR + R / MPR + p, em comparação com 0,0% no MPp + p).
Em pacientes com mieloma múltiplo recém-diagnosticado, a combinação de lenalidomida com melfalano e prednisona está associada a uma maior incidência de trombocitopenia de grau 3 e 4 (40,4% em pacientes tratados com MMR + R / MMR + p, em comparação com 13,7% em pacientes tratado com MPp + p).
• Mieloma múltiplo com pelo menos uma terapia anterior
Em pacientes com mieloma múltiplo, a combinação de lenalidomida e dexametasona está associada a uma maior incidência de neutropenia de grau 4 (5,1% dos pacientes tratados com lenalidomida / dexametasona versus 0,6% dos pacientes tratados com placebo / dexametasona Episódios de neutropenia febril de grau 4 foram observados raramente ( em 0,6% dos pacientes tratados com lenalidomida / dexametasona versus 0,0% dos pacientes tratados com placebo / dexametasona).
Em pacientes com mieloma múltiplo, a combinação de lenalidomida e dexametasona está associada a uma maior incidência de trombocitopenia grau 3 e grau 4 (em 9,9% e 1,4% dos pacientes tratados com lenalidomida / dexametasona, respectivamente. pacientes tratados com placebo / dexametasona).
• Síndromes mielodisplásicas
Em pacientes com síndromes mielodisplásicas, a lenalidomida está associada a uma maior incidência de neutropenia de grau 3 ou 4 (74,6% dos pacientes tratados com lenalidomida versus 14,9% dos pacientes tratados com placebo no estudo de Fase III). Episódios de neutropenia febril de grau 3 ou 4 foram observados em 2,2% dos pacientes tratados com lenalidomida em comparação com 0,0% dos pacientes tratados com placebo. A lenalidomida está associada a uma maior incidência de trombocitopenia de grau 3 ou 4 (37% em pacientes tratados com lenalidomida versus 1,5% em pacientes tratados com placebo no estudo de Fase III).
• Linfoma de células do manto
Em pacientes com linfoma de células do manto, a lenalidomida está associada a uma maior incidência de neutropenia de grau 3 ou 4 (43,7% dos pacientes tratados com lenalidomida em comparação com 33,7% dos pacientes no braço de controle no estudo de Fase III). Episódios de neutropenia febril de grau 3 ou 4 foram observados em 6,0% dos pacientes tratados com lenalidomida em comparação com 2,4% dos pacientes no braço de controle.
Tromboembolismo venoso
Um risco aumentado de trombose venosa profunda (TVP) e embolia pulmonar (PE) está associado ao uso de lenalidomida e dexametasona em pacientes com mieloma múltiplo e, em menor extensão, em pacientes tratados com melfalano e prednisona ou como monoterapia em pacientes com síndromes, mielodisplásico e linfoma de células do manto tratado com lenalidomida (ver secção 4.5) Nestes doentes, a administração concomitante de agentes eritropoiéticos ou história prévia de TVP pode também aumentar o risco de trombose.
Infarto do miocárdio
Foram observados casos de enfarte do miocárdio em doentes a receber lenalidomida, particularmente naqueles com fatores de risco conhecidos.
Distúrbios hemorrágicos
Os distúrbios hemorrágicos são listados em diferentes classificações com base no órgão envolvido: patologias do sistema sanguíneo e linfático; patologias do sistema nervoso (hemorragia intracraniana); patologias respiratórias, torácicas e mediastinais (epistaxe); patologias gastrointestinais (sangramento gengival, hemorragia hemorroidária, hemorragia retal); patologias renais e urinárias (hematúria); trauma, envenenamento e complicações do procedimento (contusão); patologias vasculares (equimoses).
Reações alérgicas
Foram notificados casos de reações alérgicas / hipersensibilidade. Uma possível reação cruzada entre lenalidomida e talidomida foi relatada na literatura.
Reações cutâneas graves
Casos de SSJ e NTE foram relatados. Pacientes com história prévia de erupção cutânea grave associada ao tratamento com talidomida não devem receber lenalidomida.
Segundo tumor primário
* Em ensaios clínicos em doentes com mieloma previamente tratados com lenalidomida / dexametasona versus controlos, consistindo principalmente em cancros da pele de células basais ou de células escamosas.
Leucemia mielóide aguda
• Mieloma múltiplo
Em ensaios clínicos com mieloma múltiplo recentemente diagnosticado, foram observados casos de LMA em doentes tratados com lenalidomida em combinação com melfalano ou imediatamente após melfalano em dose elevada e ASCT (ver secção 4.4). Este aumento não foi observado em ensaios clínicos em doentes com mieloma múltiplo recentemente diagnosticado tratados com lenalidomida em combinação com dexametasona em dose baixa, em comparação com talidomida em combinação com melfalano e prednisona.
• Síndromes mielodisplásicas
As variáveis basais, incluindo anormalidades citogenéticas complexas e mutação TP53, estão associadas à progressão para LMA em indivíduos dependentes de transfusão com anormalidade de deleção 5q isolada (ver seção 4.4). O risco cumulativo de progressão para LMA estimado em 2 anos foi de 13,8% em pacientes com anormalidade de deleção 5q isolada, em comparação com 17,3% para pacientes com anormalidade de deleção 5q isolada e uma "anormalidade citogenética adicional. E 38,6% em pacientes com cariótipo complexo.
Em uma "análise post-hoc de um estudo clínico conduzido com Revlimid em síndromes mielodisplásicas, a taxa de progressão estimada de 2 anos para LMA foi de 27,5% em pacientes positivos para IHC-p53 e 3,6% em pacientes positivos para IHC-p53. IHC- p53-negativo (p = 0,0038) .Em pacientes com IHC-p53-positivo, observou-se menor taxa de progressão para LMA entre aqueles que obtiveram resposta de independência transfusional (11,1%), em comparação com os que não responderam (34,8%).
Distúrbios hepáticos
Foram observadas as seguintes reações adversas pós-comercialização (frequência desconhecida): insuficiência hepática aguda e colestase (ambas com risco de vida), hepatite tóxica, hepatite citolítica, hepatite citolítica / colestática mista.
Rabdomiólise
Foram observados casos raros de rabdomiólise, alguns deles quando a lenalidomida foi administrada com uma estatina.
Distúrbios da glândula tireóide
Foram observados casos de hipotiroidismo e hipertiroidismo (ver secção 4.4 Doenças da tiróide).
Reação de exacerbação tumoral e síndrome de lise tumoral
No Estudo MCL-002, aproximadamente 10% dos pacientes tratados com lenalidomida experimentaram TFR, em comparação com 0% no braço de controle. A maioria dos eventos ocorreu no ciclo 1, todos foram considerados relacionados ao tratamento e a maioria dos relatos foi de Grau 1 ou 2. Pacientes com MIPI alto no diagnóstico e doença caracterizada por grandes massas tumorais (pelo menos uma lesão com ≥ 7 cm de diâmetro mais longo ) no início do estudo pode estar em risco de TFR. No estudo MCL-002, a TLS foi relatada para um paciente em cada um dos dois braços de tratamento. No estudo de suporte MCL-001, aproximadamente 10% dos indivíduos experimentaram TFR; todos os relatórios foram de Grau 1 ou 2 em gravidade e todos foram considerados relacionados ao tratamento. A maioria dos eventos ocorreu no ciclo 1. Não houve relatos de TLS no estudo MCL-001 (ver seção 4.4).
Problemas gastrointestinais
Perfurações gastrointestinais, que podem levar a complicações sépticas e podem estar associadas a um desfecho fatal, foram observadas durante o tratamento com lenalidomida.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas ocorridas após a autorização do medicamento é importante porque permite a monitorização contínua da relação benefício / risco do medicamento. Pede-se aos profissionais de saúde que notifiquem quaisquer suspeitas de reações adversas através do sistema nacional de notificação. No "Anexo V .
04.9 Overdose -
Não existe experiência específica no tratamento da sobredosagem com lenalidomida em doentes, embora em estudos de definição de dose alguns doentes tenham sido expostos a doses até 150 mg e, em estudos de dose única, alguns doentes tenham sido expostos a doses até 400 mg. Nestes estudos, a toxicidade limitante da dose foi essencialmente de natureza hematológica. Em caso de sobredosagem, é recomendada a terapia de suporte.
05.0 PROPRIEDADES FARMACOLÓGICAS -
05.1 "Propriedades farmacodinâmicas -
Grupo farmacoterapêutico: outros imunossupressores.
Código ATC: L04 AX04.
Mecanismo de ação
O mecanismo de ação da lenalidomida inclui propriedades antineoplásicas, antiangiogênicas, pró-eritropoiéticas e imunomoduladoras. Especificamente, a lenalidomida inibe a proliferação de células tumorais hematopoiéticas específicas (incluindo células plasmáticas cancerosas do MM e aquelas com deleção do cromossomo 5)., aumenta a imunidade mediada por células T e células assassinas naturais (NK) e aumenta o número de células NKT; inibe a angiogênese, bloqueando a migração e adesão de células endoteliais e a formação de microvasos; aumenta a produção de hemoglobina fetal pelas células-tronco hematopoéticas CD34 + e inibe a produção de citocinas pró-inflamatórias (por exemplo, TNF-α e IL-6) pelos monócitos.
Em MDS com anomalia de deleção 5q isolada, a lenalidomida demonstrou inibir seletivamente o clone anormal, aumentando a apoptose das células Del (5q).
A lenalidomida se liga diretamente ao cereblon, um componente de um complexo de enzima cullin-RING E3 ubiquitina ligase, que inclui a proteína 1 de ligação ao ácido desoxirribonucléico (DNA) (DDB1,Proteína-1 de ligação a danos de DNA), descarte 4 (CUL4) e regulador de descarte 1 (Roc1). Na presença da lenalidomida, o cereblon liga-se às proteínas substrato Aiolos e Ikaros, que são fatores de transcrição linfoide, causando sua ubiquitinação e posterior degradação, com conseqüente efeito citotóxico e imunomodulador.
Eficácia clínica e segurança
A lenalidomida foi avaliada em dois estudos de Fase III em mieloma múltiplo recém-diagnosticado e em dois estudos de Fase III em mieloma múltiplo refratário recidivante, conforme descrito abaixo.
Mim E eu ou m um múltiplo recém-diagnosticado
Lenalidomida em combinação com dexametasona em pacientes não elegíveis para transplante de células-tronco
A eficácia e segurança da lenalidomida foram avaliadas em um estudo multicêntrico, randomizado, aberto, de três braços de Fase III (MM-020) em pacientes com 65 anos de idade ou mais ou, se menores de 65 anos, que foram inelegíveis para o transplante de células-tronco devido à decisão do paciente ou indisponibilidade do transplante de células-tronco por custo ou outros motivos. O estudo (MM-020) comparou lenalidomida e dexametasona (Rd) administradas por 2 durações de tratamento diferentes (por exemplo, até a progressão da doença [braço Rd] ou até dezoito ciclos de 28 dias [72 semanas, braço Rd18]) com melfalano, prednisona e talidomida (MPT) por até doze ciclos de 42 dias (72 semanas). Os pacientes foram randomizados (1: 1: 1) para um dos 3 braços de tratamento. Na randomização, os pacientes foram estratificados por idade (≤ 75 anos vs> 75 anos), estágio (ISS estágios I e II vs estágio III) e país.
Os pacientes nos braços Rd e Rd18 receberam lenalidomida 25 mg uma vez ao dia nos dias 1 a 21 dos ciclos de 28 dias, de acordo com o braço do protocolo. Dexametasona 40 mg foi administrado uma vez ao dia nos Dias 1, 8, 15 e 22 de cada ciclo de tratamento de 28 dias. A dose inicial e o regime para Rd e Rd18 foram ajustados para idade e função renal (ver seção 4.2). Pacientes com> 75 anos de idade receberam uma dose de dexametasona de 20 mg uma vez ao dia nos Dias 1, 8, 15 e 22 de cada 28 dias ciclo de tratamento Todos os pacientes foram submetidos à profilaxia anticoagulante (heparina de baixo peso molecular, varfarina, heparina, aspirina em baixa dosagem) durante o estudo.
O endpoint primário de eficácia no estudo foi a sobrevida livre de progressão (Progressão Sobrevivência Grátis, PFS). No total, 1.623 pacientes foram incluídos no estudo: 535 pacientes randomizados para Rd, 541 pacientes randomizados para Rd18 e 547 pacientes randomizados para MPT. As características demográficas e relacionadas à doença dos pacientes no início do estudo foram bem equilibradas em todos os 3 braços.No geral, os participantes do estudo tinham doença avançada: da população total do estudo, 41% estavam no estágio III do ISS, 9% tinham insuficiência renal grave (depuração da creatinina [CLcr]
Em uma análise atualizada de PFS, PFS2 e sobrevida global (OS) usando a data de corte de 3 de março de 2014, onde o tempo médio de acompanhamento para todos os indivíduos sobreviventes foi de 45,5 meses, os resultados do estudo são apresentados em Tabela 5.
Tabela 5: Resumo dos dados gerais de eficácia
AMT = terapia anti-mieloma; IC = intervalo de confiança; CR = resposta completa; d = dexametasona em dose baixa; HR = razão de risco; IMWG = Grupo de Trabalho Internacional de Mieloma; IRAC = Comitê de Adjudicação de Resposta Independente; M = melfalano; max = máximo; min = mínimo; NS = não estimável; OS = sobrevida global; P = prednisona; PFS = sobrevivência livre de progressão; PR = resposta parcial; R = lenalidomida; Rd = Rd administrado até a progressão da doença documentada; Rd18 = Rd administrado por ≥ 18 ciclos; SE = erro padrão; T = talidomida; VGPR = resposta parcial ótima; vs = versus.
a A mediana é baseada na estimativa de Kaplan-Meier.
b IC 95% sobre a mediana.
c Com base no modelo de riscos proporcionais de Cox comparando as funções de risco associadas aos braços de tratamento indicados.
d O valor p é baseado no teste de log rank não estratificado das diferenças nas curvas de Kaplan-Meier entre os braços de tratamento indicados.
e Endpoint exploratório (PFS2)
f A mediana é a estatística univariada sem correção de truncamento.
g Melhor avaliação da resposta adjudicada durante a fase de tratamento do estudo (para as definições de cada categoria de resposta Data limite de dados = 24 de maio de 2013).
h Data limite = 24 de maio de 2013
Lenalidomida em combinação com melfalano e prednisona, seguida por monoterapia de manutenção, em pacientes não elegíveis para transplante
A segurança e eficácia da lenalidomida (MPR) foram avaliadas em um estudo multicêntrico, randomizado, duplo-cego, de Fase III de 3 braços (MM-015) em pacientes com 65 anos de idade ou mais e com creatinina sérica. 75 anos) e estágio (ISS estágios I e II vs estágio III).
Este estudo examinou o uso da terapia combinada MMR (melfalan 0,18 mg / kg por via oral nos dias 1-4 de ciclos repetidos de 28 dias; prednisona 2 mg / kg por via oral nos dias 1-4 de ciclos repetidos de 28 dias; e lenalidomida 10 mg / dia por via oral nos dias 1-21 de ciclos repetidos de 28 dias), para terapia de indução, até um máximo de 9 ciclos. Pacientes que completaram 9 ciclos, ou que não conseguiram completar os 9 ciclos devido à intolerância, mudaram para monoterapia de manutenção, iniciada com lenalidomida 10 mg por via oral nos dias 1-21 de ciclos repetidos de 28 dias, até a progressão da doença
O endpoint primário de eficácia no estudo foi a sobrevida livre de progressão (PFS). No total, 459 pacientes foram incluídos no estudo: 152 pacientes randomizados para MMR + R, 153 pacientes randomizados para MMR + p e 154 pacientes randomizados para MPp + p . As características demográficas e relacionadas à doença do paciente no início do estudo estavam bem equilibradas em todos os 3 braços; em particular, aproximadamente 50% dos pacientes inscritos em cada braço tinham as seguintes características: ISS estágio III e depuração da creatinina
Em uma "análise de PFS, PFS2 e OS usando a data de corte de abril de 2013, para a qual o tempo médio de acompanhamento para todos os indivíduos sobreviventes foi de 62,4 meses, os resultados do estudo são apresentados na Tabela 6..
Tabela 6: Resumo dos dados gerais de eficácia
IC = intervalo de confiança; CR = resposta completa; HR = razão de risco; M = melfalano; NS = não estimável; OS = sobrevida global; p = placebo; P = prednisona;
PD = doença progressiva; PR = resposta parcial; R = lenalidomida; SD = doença estável; VGPR = resposta parcial ótima.
a A mediana é baseada na estimativa de Kaplan-Meier.
¤ PFS2 (um desfecho exploratório) foi definido para todos os pacientes (ITT) como o tempo desde a randomização até o início da terapia anti-mieloma de terceira linha ou morte por qualquer causa, para todos os pacientes randomizados
Estudos de suporte em mieloma múltiplo recém-diagnosticado
Um estudo aberto, randomizado, multicêntrico de Fase III (ECOG E4A03) foi conduzido em 445 pacientes com mieloma múltiplo recém-diagnosticado; 222 pacientes foram randomizados para o braço da lenalidomida / dexametasona em dose baixa e 223 foram randomizados para o braço da lenalidomida / dexametasona em dose padrão. Os pacientes randomizados no braço da lenalidomida / dose padrão de dexametasona receberam lenalidomida 25 mg / dia nos dias 1 a 21, a cada 28 dias, mais dexametasona 40 mg / dia nos dias 1 a 4, 9 a 12 e 17 aos 20, a cada 28 dias, para os primeiros quatro ciclos. Os pacientes randomizados para o braço de lenalidomida / dexametasona em dose baixa receberam lenalidomida 25 mg / dia nos dias 1 a 21, a cada 28 dias, mais dexametasona em dose baixa 40 mg / dia nos dias 1, 8, 15 e 22, a cada 28 dias . No grupo da lenalidomida / dexametasona em dose baixa, 20 pacientes (9,1%) tiveram pelo menos uma interrupção da dose em comparação com 65 pacientes (29,3%) no braço da lenalidomida / dexametasona em dose padrão.
Em uma análise post-hoc, a mortalidade mais baixa foi observada no braço de lenalidomida / dexametasona em dose baixa 6,8% (15/220), em comparação com o braço de lenalidomida / dose padrão de dexametasona 19,3% (43/223), no braço de dexametasona recém-diagnosticado população de pacientes com mieloma, com seguimento médio de 72,3 semanas.
No entanto, com acompanhamento prolongado, a diferença na sobrevida global em favor de lenalidomida / dexametasona em dose baixa tende a diminuir.
Mieloma múltiplo com pelo menos uma terapia anterior
A eficácia e segurança da lenalidomida foram avaliadas em dois estudos multicêntricos de Fase III, randomizados, duplo-cegos, controlados com placebo, grupos paralelos (MM-009 e MM-010) de lenalidomida em combinação com monoterapia de dexametasona versus Dexametasona em pacientes previamente tratados com mieloma múltiplo. Dos 353 pacientes incluídos nos estudos MM-009 e MM-010 tratados com lenalidomida / dexametasona, 45,6% tinham 65 anos de idade ou mais. Dos 704 pacientes avaliados nos estudos MM-009 e MM-010, 44,6% tinham 65 anos ou mais.
Em ambos os estudos, os pacientes no grupo lenalidomida / dexametasona (len / des) receberam lenalidomida 25 mg por via oral uma vez ao dia nos dias 1 a 21 e uma cápsula de placebo de aparência idêntica uma vez ao dia. Dias 22 a 28 de cada ciclo de 28 dias. O grupo placebo / dexametasona (placebo / des) tomou 1 cápsula de placebo nos dias 1 a 28 de cada ciclo de 28 dias. Os pacientes em ambos os grupos tomaram 40 mg de dexametasona por via oral uma vez ao dia nos dias 1 a 4, 9 a 12 e
17 a 20 de cada ciclo de 28 dias, durante os primeiros 4 ciclos de terapia. Após os primeiros 4 cursos de terapia, a dose de dexametasona foi reduzida para 40 mg por via oral uma vez ao dia nos dias 1 a 4 de cada ciclo de 28 dias. Em ambos os estudos, o tratamento deveria continuar até a progressão da doença. Ajustes de dosagem foram permitidos em ambos os estudos com base em resultados clínicos e laboratoriais.
O endpoint primário de eficácia em ambos os estudos foi o tempo para a progressão da doença (TTP, hora de progressão) Um total de 353 pacientes foram avaliados no estudo MM-009: 177 no grupo lenalidomida / dexametasona e 176 no grupo placebo / dexametasona. Um total de 351 pacientes foram avaliados no estudo MM-010: 176 no grupo lenalidomida / dexametasona e 175 no grupo placebo / dexametasona.
Em ambos os estudos, os grupos lenalidomida / dexametasona e placebo / dexametasona tinham características demográficas iniciais e relacionadas à doença comparáveis. Ambas as populações de pacientes tinham uma idade mediana de 63 anos, com uma proporção comparável entre pacientes do sexo masculino e feminino.Grupo de Oncologia Cooperativa Oriental), tanto o número quanto o tipo de terapias anteriores eram comparáveis em ambos os grupos.
Análises provisório pré-programado para ambos os estudos mostrou que a terapia combinada de lenalidomida / dexametasona mostrou uma melhora estatisticamente significativa (p
Uma análise de eficácia de acompanhamento estendida foi conduzida por uma mediana de 130,7 semanas. A Tabela 7 mostra os resultados das análises de eficácia de acompanhamento - estudos conjuntos MM-009 e MM-010.
Nesta análise agrupada de acompanhamento estendido, o TTP mediano foi de 60,1 semanas (IC de 95%: 44,3, 73,1) em pacientes tratados com lenalidomida / dexametasona (N = 353) em comparação com a mediana de 20, 1 semana (IC de 95%: 17,7, 20,3) em pacientes tratados com placebo / dexametasona (N = 351). A sobrevida livre de progressão mediana foi de 48,1 semanas (IC 95%: 36,4, 62,1) em pacientes tratados com lenalidomida / dexametasona em comparação com um tempo médio de 20,0 semanas (IC 95%: 16, 1, 20,1) em pacientes tratados com placebo / dexametasona . A duração média do tratamento foi de 44,0 semanas (min: 0,1, máx: 254,9) para lenalidomida / dexametasona e 23,1 semanas (min: 0,3, máx: 238,1) para placebo / dexametasona. Em ambos os estudos, as taxas de resposta completa (CR, resposta completa), resposta parcial (PR, resposta parcial) e a resposta geral (CR + PR) no grupo lenalidomida / dexametasona permaneceu significativamente maior do que no grupo dexametasona / placebo. A sobrevida global mediana na análise de acompanhamento estendido dos estudos conjuntos é 164,3 semanas (IC 95%: 145,1, 192,6) em pacientes tratados com lenalidomida / dexametasona em comparação com 136,4 semanas (IC 95%: 113,1, 161,7) em pacientes tratados com placebo / dexametasona. Apesar do fato de que 170 dos 351 pacientes randomizados para tratamento com placebo / dexametasona receberam terapia com lenalidomida após a progressão da doença ou após cegueira, a análise de sobrevida global combinada demonstrou uma vantagem de sobrevida estatisticamente significativa para o grupo lenalidomida / dexametasona em comparação com o placebo / grupo dexametasona (razão de risco = 0,833, IC 95% = [0,687, 1,009], p = 0,045).
Tabela 7: Resumo dos resultados das análises de eficácia na data limite para acompanhamento estendido - Estudos conjuntos MM-009 e MM-010 (respectivas datas limite: 23 de julho de 2008 e 2 de março de 2008)
a: Análise univariada bilateral comparando as curvas de sobrevivência entre os grupos de tratamento. b: Teste qui-quadrado bicaudal com correção de continuidade.
Síndromes mielodisplásicas
A eficácia e segurança da lenalidomida foram avaliadas em pacientes com anemia dependente de transfusão devido a síndromes mielodisplásicas de risco baixo ou intermediário-1 associadas a anormalidade citogenética de deleção 5q, com ou sem outras anormalidades citogenéticas, em dois estudos principais.: A Fase III, multicêntrico , estudo randomizado, duplo-cego, controlado por placebo, lenalidomida oral de 3 braços, duas doses (10 mg e 5 mg) versus placebo (MDS-004); e um estudo de Fase II, multicêntrico, braço único, rótulo aberto, em lenalidomida (10 mg) (MDS-003).
Os resultados abaixo representam a população com intenção de tratar estudada em MDS-003 e MDS-004; os resultados para a subpopulação com deleção 5q isolada são apresentados separadamente (ver secção 4.1 para a indicação aprovada).
No estudo MDS-004, no qual 205 pacientes foram igualmente randomizados para tratamento com lenalidomida 10 mg, 5 mg ou placebo, a análise de eficácia primária consistiu na comparação das taxas de resposta de independência de transfusão nos braços de lenalidomida de 10 mg e 5 mg em comparação com o placebo. braço (fase duplo-cega de 16 a 52 semanas e fase aberta até um total de 156 semanas). Em pacientes que não apresentaram pelo menos uma resposta eritróide leve após 16 semanas, o tratamento foi interrompido. pacientes que tinham evidência de pelo menos uma resposta eritróide leve pode continuar a terapia até a recorrência eritróide, progressão da doença ou toxicidade inaceitável. após 16 semanas de tratamento, eles foram autorizados a mudar de placebo para 5 mg de lenalidomida ou continuar com tratamento com lenalidomida em dose mais elevada (5 mg a 10 mg).
No estudo MDS-003, no qual 148 pacientes foram tratados com lenalidomida em uma dose de 10 mg, a análise de eficácia primária consistiu em uma avaliação da eficácia dos tratamentos com lenalidomida na obtenção de melhora hematopoiética em indivíduos com síndromes mielodisplásicas. 1
Tabela 8: Resumo dos resultados de eficácia - MDS-004 (fase duplo-cega) e MDS-003, população com intenção de tratar
† Indivíduos tratados com lenalidomida 10 mg em 21 do ciclo de 28 dias
†† Indivíduos tratados com lenalidomida 5 mg em 28 do ciclo de 28 dias
* A maioria dos pacientes do grupo placebo descontinuou o tratamento duplo-cego devido à falta de eficácia após 16 semanas de tratamento, antes de entrar na fase de rótulo aberto
# Associado a um aumento em Hgb de ≥ 1 g / dL
∞ Não alcançado (mediana não alcançada)
No estudo MDS-004, uma proporção significativamente maior de pacientes com síndromes mielodisplásicas atingiu o desfecho primário de independência de transfusão (> 182 dias) com lenalidomida 10 mg, em comparação com placebo (55,1% vs. 6,0%) entre os 47 pacientes com anormalidade citogenética .
Del (5q) isolado e tratado com 10 mg de lenalidomida, 27 pacientes (57,4%) alcançaram independência das transfusões de eritrócitos.
O tempo médio para a independência da transfusão no braço da lenalidomida 10 mg foi de 4,6 semanas. A duração mediana da independência da transfusão não foi alcançada em nenhum dos braços de tratamento, mas espera-se que exceda 2 anos para os indivíduos tratados. Com lenalidomida. O aumento médio em a hemoglobina (Hgb) da linha de base no braço de 10 mg foi de 6,4 g / dL.
Os desfechos adicionais do estudo incluíram resposta citogenética (respostas citogenéticas maiores e menores foram observadas em 30,0% e 24,0% dos indivíduos no braço de 10 mg, respectivamente), avaliação da qualidade de vida relacionada à saúde (QVRS) e progressão para leucemia mieloide aguda. Os resultados da resposta citogenética e da QVRS foram consistentes com os resultados do endpoint primário e a favor do tratamento com lenalidomida em vez do placebo.
No estudo MDS-003, uma alta porcentagem de pacientes com síndromes mielodisplásicas alcançou independência de transfusão (> 182 dias) com lenalidomida 10 mg (58,1%). O tempo médio para a independência transfusional foi de 4,1 semanas, a duração mediana da independência transfusional foi de 114,4 semanas e o aumento médio da hemoglobina (Hgb) foi de 5,6 g / dl.
Respostas citogenéticas maiores e menores foram observadas em 40,9% e 30,7% dos indivíduos, respectivamente.
Uma grande porcentagem de indivíduos inscritos no MDS-003 (72,9%) e no MDS-004 (52,7%) haviam sido tratados anteriormente com agentes estimuladores da eritropoiese.
Linfoma de células do manto
A eficácia e segurança da lenalidomida em pacientes com linfoma de células do manto foram avaliadas em um estudo de Fase II, multicêntrico, randomizado, aberto versus a escolha do investigador de monoterapia em pacientes que eram refratários ao último regime ou que apresentaram de uma a três recidivas ( Estudo MCL-002).
Pacientes com pelo menos 18 anos de idade com linfoma de células do manto confirmado histologicamente e doença mensurável na TC foram inscritos. Os pacientes deveriam ter recebido tratamento prévio adequado com pelo menos um regime de quimioterapia de combinação anterior. Além disso, os pacientes deveriam ser inelegíveis para quimioterapia intensiva e / ou transplante na inclusão do estudo. Os pacientes foram randomizados 2: 1 para lenalidomida ou o braço de controle. A escolha do investigador do tratamento foi decidida mais cedo. De randomização e consistia em monoterapia com clorambucil, citarabina, rituximabe, fludarabina ou gencitabina.
A lenalidomida foi administrada por via oral, na dose de 25 mg uma vez ao dia durante os primeiros 21 dias (G1 a G21) de ciclos repetidos de 28 dias até progressão ou toxicidade inaceitável. Os pacientes com insuficiência renal moderada deveriam receber uma dose inicial mais baixa de lenalidomida (10 mg por dia) com o mesmo esquema.
Os dados demográficos da linha de base foram comparáveis entre a lenalidomida e os braços de controle. Ambas as populações de pacientes tinham uma idade mediana de 68,5 anos, com uma proporção comparável de pacientes do sexo masculino para feminino.O status de desempenho do ECOG e o número de terapias anteriores eram comparáveis em ambos os grupos.
O endpoint primário de eficácia em MCL-002 foi a sobrevivência livre de progressão (PFS).
Os resultados de eficácia para a população com intenção de tratar (ITT) foram avaliados pela Comissão de Revisão Independente (Comitê de Revisão Independente, IRC) e são apresentados na tabela abaixo.
Tabela 9: Resumo dos resultados de eficácia - estudo MCL-002, população com intenção de tratar
IC = intervalo de confiança; CRR = taxa de resposta completa; CR = resposta completa; CRu = resposta completa não confirmada; DMC = comitê de monitoramento de dados; ITT = intenção de tratar; HR = razão de risco; KM = Kaplan-Meier; MIPI = Índice Prognóstico Internacional de Linfoma de Células do Manto; NP = não relevante; ORR = taxa de resposta geral; PD = doença progressiva; PFS = sobrevivência livre de progressão; PR = resposta parcial; SCT = transplante de células-tronco; SD = doença estável; SE = erro padrão.
a A mediana é baseada na estimativa KM.
b O intervalo foi calculado como IC de 95% em relação ao tempo médio de sobrevivência.
e A média e a mediana são as estatísticas univariadas sem correção de truncamento.
d As variáveis de estratificação incluíram o tempo desde o diagnóstico até a primeira dose (
O teste sequencial foi baseado em uma média ponderada da estatística do teste de log rank, usando o teste de log rank não estratificado para aumento do tamanho da amostra e o teste de log rank não estratificado da análise primária. Os pesos são baseados nos eventos observados na data da terceira reunião do RCD e são baseados na diferença entre os eventos observados e os eventos esperados no momento da análise primária.
A FC sequencial associada e o IC 95% correspondente são apresentados.
No estudo MCL-002 na população ITT, houve um aumento geral aparente de mortes em 20 semanas no braço da lenalidomida, 22/170 (13%), em comparação com 6/84 (7%) no braço controle. Em doentes com elevada carga tumoral, os valores correspondentes foram 16/81 (20%) e 2/28 (7%) (ver secção 4.4).
População pediátrica
A Agência Europeia de Medicamentos dispensou a obrigação de apresentação dos resultados dos estudos com Revlimid em todos os subgrupos da população pediátrica para mieloma múltiplo, síndromes mielodisplásicas e linfoma de células do manto (ver secção 4.2 para informação sobre “utilização pediátrica).
05.2 "Propriedades farmacocinéticas -
A lenalidomida tem um átomo de carbono assimétrico; portanto, sua molécula existe nas formas opticamente ativas S (-) e R (+). A lenalidomida é produzida como uma mistura racêmica. A lenalidomida é geralmente mais solúvel em solventes orgânicos, mas exibe solubilidade máxima em solução de HCl 0,1N.
Absorção
A lenalidomida é rapidamente absorvida após administração oral em voluntários saudáveis, em jejum, atingindo concentrações plasmáticas máximas entre 0,5 e 2 horas após a administração. Em pacientes e voluntários saudáveis, a concentração máxima (Cmax) el "área sob a curva de concentração-tempo ( A AUC) aumenta proporcionalmente com o aumento da dose. Doses repetidas não causam acumulação significativa do fármaco. No plasma, a concentração relativa dos enantiómeros S e R da lenalidomida é de aproximadamente 56% e 44%, respetivamente.
A co-administração de uma refeição hipercalórica e rica em gorduras em voluntários saudáveis reduz a extensão da absorção, resultando numa diminuição de aproximadamente 20% na área abaixo da AUC e numa diminuição de 50% na Cmax plasmática. No entanto, nos estudos principais de registro de mieloma múltiplo e síndromes mielodisplásicas, nos quais a segurança e eficácia da lenalidomida foram estabelecidas, o medicamento foi administrado sem levar em consideração a ingestão de alimentos. Portanto, a lenalidomida pode ser administrada com ou sem alimentos.
As análises farmacocinéticas populacionais indicam que a taxa de absorção da lenalidomida oral é semelhante entre pacientes com mieloma múltiplo, pacientes com síndromes mielodisplásicas e pacientes com linfoma de células do manto.
Distribuição
Em vitro, A lenalidomida marcada com 14C liga-se fracamente às proteínas plasmáticas, com um valor médio de 23% e 29%, respectivamente, em pacientes com mieloma múltiplo e voluntários saudáveis.
A lenalidomida está presente no sêmen (
Biotransformação e eliminação
Os resultados dos estudos de metabolismo humano realizados em vitro indicam que a lenalidomida não é metabolizada pelas enzimas do citocromo P450, sugerindo que a administração de lenalidomida juntamente com medicamentos que inibem as enzimas do citocromo P450 é improvável que produza interações medicamentosas metabólicas em humanos. em vitro indicam que a lenalidomida não tem efeito inibidor em CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A ou UGT1A1. Portanto, é improvável que a lenalidomida cause interações medicamentosas clinicamente relevantes quando administrada concomitantemente com substratos dessas enzimas.
Educação em vitro indicam que a lenalidomida não é um substrato da proteína de resistência ao câncer de mama humano (BCRP), transportadores de proteínas de resistência a múltiplas drogas (MRP) MRP1, MRP2 ou MRP3, transportadores de ânions orgânicos (OAT) OAT1 e OAT3, transportador de ânions orgânicos polipeptídicos (OATP) OATP1B1, orgânicos transportadores catiônicos (OCT) OCT1 e OCT2, proteína de extrusão de drogas e toxinas (MATE) MATE1 e novos transportadores catiônicos orgânicos (OCTN) OCTN1 e OCTN2.
Educação em vitro indicam que a lenalidomida não tem efeito inibidor na bomba de exportação de sais biliares humanos (BSEP), BCRP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3 e OCT2.
A maior parte da lenalidomida é eliminada por excreção urinária A contribuição da excreção renal para a depuração total em indivíduos com função renal normal foi de 90%, enquanto 4% da lenalidomida foi eliminada nas fezes.
A lenalidomida é fracamente metabolizada, pelo que 82% da dose é excretada na forma inalterada na urina. A hidroxilenalidomida e a N-acetil-lenalidomida representam 4,59% e 1,83% da dose excretada, respetivamente. A depuração renal da lenalidomida excede a taxa de filtração glomerular, portanto, pelo menos em certa medida, é secretada ativamente.
Em doses de 5 a 25 mg / dia, a meia-vida plasmática é de aproximadamente 3 horas em voluntários saudáveis e varia de 3 a 5 horas em pacientes com mieloma múltiplo, síndromes mielodisplásicas ou linfoma de células do manto.
Pacientes idosos
Não foram realizados estudos clínicos específicos para avaliar a farmacocinética da lenalidomida em pacientes idosos. As análises farmacocinéticas populacionais incluíram pacientes com idade entre 39 e 85 anos e indicaram que a idade não afeta a depuração (concentração plasmática) da lenalidomida. Como os pacientes idosos são mais propensos a ter função renal diminuída, recomenda-se cautela na seleção da dose e a monitorização da função renal é aconselhada como medida de precaução.
Falência renal
A farmacocinética da lenalidomida foi estudada em indivíduos com insuficiência renal causada por doenças não malignas. Neste estudo, foram utilizados dois métodos para a classificação da função renal: depuração da creatinina urinária medida ao longo de 24 horas e depuração da creatinina estimada com a fórmula de Cockcroft-Gault. Os resultados indicam que, à medida que a função renal diminui (a meia-vida da lenalidomida aumentou de aproximadamente 3,5 horas em indivíduos com depuração da creatinina> 50 ml / min a mais de 9 horas em indivíduos com função renal comprometida
Insuficiência Hepática
As análises farmacocinéticas da população incluíram pacientes com insuficiência hepática leve (N = 16, bilirrubina total> 1 a ≤ 1,5 x ULN (Limite Superior do Normal) ou AST> LSN) e indicam que "insuficiência hepática leve não afeta a depuração (concentração plasmática) de lenalidomida Não existem dados disponíveis em doentes com compromisso hepático moderado a grave.
Outros fatores intrínsecos
As análises farmacocinéticas da população indicam que o peso corporal (33-135 kg), sexo, raça e tipo de malignidade hematológica (MM, MDS ou MCL) não têm efeito clinicamente relevante na depuração da lenalidomida em doentes adultos.
05.3 Dados de segurança pré-clínica -
Um estudo de desenvolvimento embriofetal foi conduzido em macacos tratados com lenalidomida em doses variando de 0,5 a 4 mg / kg / dia. Os resultados deste estudo indicaram que a lenalidomida causa malformações externas, incluindo orifício anal não patente e malformações das extremidades superiores e inferiores (partes das extremidades que são curvas, encurtadas, malformadas, mal-giradas e / ou ausentes, oligo e / ou polidactilia) na prole de macacas que receberam a droga durante a gestação.
Também foram observados diferentes efeitos viscerais em fetos individuais (descoloração, focos vermelhos em vários órgãos, pequena massa incolor acima da válvula atrioventricular, vesícula biliar pequena, diafragma malformado).
A lenalidomida apresenta um risco potencial de toxicidade aguda; em roedores, as doses letais mínimas após a administração oral foram> 2.000 mg / kg / dia. A administração oral repetida de 75, 150 e 300 mg / kg / dia por até 26 semanas resultou em um aumento reversível relacionado ao tratamento na mineralização pélvica renal em ratos, principalmente mulheres, em todos os níveis de dosagem. O nível de efeitos adversos não observáveis (NOAEL, nenhum nível de efeito adverso observado) foi considerado inferior a 75 mg / kg / dia e é aproximadamente 25 vezes superior à exposição humana diária com base nos valores de AUC. Em macacos, a administração oral repetida de 4 e 6 mg / kg / dia por períodos de até 20 semanas produziu mortalidade e toxicidade significativas (perda de peso acentuada, contagem de glóbulos brancos reduzida, glóbulos vermelhos e plaquetas, hemorragias em vários órgãos, inflamação de trato gastrointestinal, atrofia do tecido linfático e da medula óssea). Também em macacos, a administração oral repetida de 1 e 2 mg / kg / dia, por períodos de até 1 ano, produziu alterações reversíveis na celularidade da medula óssea, uma ligeira redução na proporção de células mielo-eritróides e atrofia do timo. Foi observada uma ligeira diminuição na contagem de glóbulos brancos para 1 mg / kg / dia, o que corresponde aproximadamente à mesma dose em humanos, com base na comparação da AUC.
Estudos de mutagenicidade realizados em vitro (mutação bacteriana, linfócitos humanos, linfoma murino, transformação em células embrionárias de hamster sírio) e na Vivo (teste do micronúcleo do rato) não revelou quaisquer efeitos relacionados com o fármaco ao nível do gene ou ao nível do cromossoma. Não foram realizados estudos de carcinogenicidade com lenalidomida.
A toxicidade para o desenvolvimento foi previamente estudada em coelhos. Nestes estudos, os coelhos receberam por via oral 3, 10 e 20 mg / kg / dia de lenalidomida. Ausência do lobo intermediário do pulmão foi observada na dose de 10 e 20 mg / kg / dia, com correlação com a dose, e rim ectópico na dose de 20 mg / kg / dia, embora essas condições tenham sido observadas na dose de 20 mg / kg / dia, doses tóxicas para a mãe, podem ser atribuídas a um efeito direto. Em doses de 10 e 20 mg / kg / dia, alterações nos tecidos moles e esqueleto também foram observadas em fetos.
06.0 INFORMAÇÕES FARMACÊUTICAS -
06.1 Excipientes -
Conteúdo da cápsula
Lactose anidra
Celulose microcristalina
Croscarmelose de sódio
Estearato de magnesio
Concha da cápsula
Geléia
Dióxido de titânio (E171)
Indigo carmim (E132)
Óxido de ferro amarelo (E172)
Tinta da redação
Goma laca
Propileno glicol
Óxido de ferro preto (E172)
Hidróxido de potássio
06.2 Incompatibilidade "-
Não é relevante.
06.3 Período de validade "-
3 anos.
06.4 Precauções especiais de armazenamento -
Este medicamento não requer quaisquer condições especiais de armazenamento.
06.5 Natureza da embalagem primária e conteúdo da embalagem -
Cloreto de polivinila (PVC) / policlorotrifluoroetileno (PCTFE) / blister de folha de alumínio contendo 7 cápsulas.
Embalagem com 21 cápsulas.
06.6 Instruções de uso e manuseio -
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA "AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO" -
Celgene Europe Limited
1 Longwalk Road
Stockley Park
Uxbridge
UB11 1DB
Reino Unido
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO -
EU / 1/07/391/002
038016022
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO -
Data da primeira autorização: 14 de junho de 2007
Data da renovação mais recente: 14 de junho de 2012
10.0 DATA DE REVISÃO DO TEXTO -
D.CCE setembro de 2016
11.0 PARA RADIOFármacos, PREENCHA OS DADOS NA DOSIMETRIA DE RADIAÇÃO INTERNA -
12.0 PARA MEDICAMENTOS DE RÁDIO, INSTRUÇÕES MAIS DETALHADAS SOBRE PREPARAÇÃO EXTEMPORÁRIA E CONTROLE DE QUALIDADE -